Difference between revisions of "20.109(F20):M2D2"
Noreen Lyell (Talk | contribs) (→Introduction) |
Noreen Lyell (Talk | contribs) |
||
| Line 18: | Line 18: | ||
[[Image:Fa20 M2D1 protein purification.png|thumb|550px|center|'''Schematic of affinity separation process.''' For purification, agarose beads (yellow) are coated with nickel (green). When cell lysate is added to the nickel-coated agarose beads, His-tagged protein of interest (blue) adheres to the beads and other proteins in the lysate (orange) are washed from the beads.]] | [[Image:Fa20 M2D1 protein purification.png|thumb|550px|center|'''Schematic of affinity separation process.''' For purification, agarose beads (yellow) are coated with nickel (green). When cell lysate is added to the nickel-coated agarose beads, His-tagged protein of interest (blue) adheres to the beads and other proteins in the lysate (orange) are washed from the beads.]] | ||
| + | |||
| + | ==Protocols== | ||
| + | ===Part 1: Electrophorese confirmation digests=== | ||
| + | |||
| + | Electrophoresis is a technique that separates large molecules by size using an applied electrical field and a sieving matrix. DNA, RNA and proteins are the molecules most often studied with this technique; agarose and acrylamide gels are the two most common sieves. The molecules to be separated enter the matrix through a well at one end and are pulled through the matrix when a current is applied across it. The larger molecules get entwined in the matrix and are stalled; the smaller molecules wind through the matrix more easily and travel farther away from the well. The distance a DNA fragment travels is inversely proportional to the log of its length. Over time fragments of similar length accumulate into “bands” in the gel. Higher concentrations of agarose can be used to resolve smaller DNA fragments. | ||
| + | |||
| + | [[Image:Mod1 2 photo.jpg|thumb|right|350px|'''Diagram of gel electrophoresis chamber.''' Larger sized DNA molecules will remain close to the well where the sample was loaded and smaller DNA molecules will migrate through the gel toward the positive electrode.]] | ||
| + | |||
| + | DNA and RNA are negatively charged molecules due to their phosphate backbone, and they naturally travel toward the positive electrode at the far end of the gel. Today you will separate DNA fragments using an agarose matrix. Agarose is a polymer that comes from seaweed. To prepare these gels, agarose and 1X TAE buffer (Tris base, acetic acid, and EDTA) are microwaved until the agarose is melted and fully dissolved. The molten agar is then poured into a horizontal casting tray, and a comb is added. Once the agar has solidified, the comb is removed, leaving wells into which the DNA samples can be loaded. | ||
| + | |||
| + | You will use a 1% agarose gel with SYBR Safe DNA stain (prepared by the teaching faculty) to separate the DNA fragments in your four digested samples as well as a reference lane of molecular weight markers (also called a DNA ladder). | ||
| + | |||
| + | #Add 5 μL of 6x loading dye to the digests.[[Image:Mod1 2 agaro gel.jpg|thumb|right|200px|'''Illustration of proper gel loading technique.''']] | ||
| + | #*Loading dye contains bromophenol blue as a tracking dye to follow the progress of the electrophoresis (so you don’t run the smallest fragments off the end of your gel!) as well as glycerol to help the samples sink into the wells. | ||
| + | #Flick the eppendorf tubes to mix the contents, then quick spin them in the microfuge to bring the contents of the tubes to the bottom. | ||
| + | #Load 25 μL of each digest into the gel, as well as 10 μL of 1kb DNA ladder. | ||
| + | #*Be sure to record the order in which you load your samples! | ||
| + | #*To load your samples, draw the volume listed above into the tip of your P200 or P20. Lower the tip below the surface of the buffer and directly over the well. Avoid lowering the tip too far into the well itself so as to not puncture the well. Expel your sample slowly into the well. Do not release the pipet plunger until after you have removed the tip from the gel box (or you'll draw your sample back into the tip!). | ||
| + | #Once all the samples have been loaded, attach the gel box to the power supply and electrophorese the gel at 125 V for 45 minutes. | ||
| + | |||
| + | ===Part 2: Lyse BL21-A1 pET_MBP_SNAP_TDP43-RRM12 cells=== | ||
| + | Previously, the teaching faculty inoculated 5 mL of LB media containing kanamycin with BL21-A1 pET_MBP_SNAP_TDP43-RRM12, incubated the culture at 37 °C overnight. The next day the overnight bacterial culture was diluted 1:10 in 50 mL total of fresh LB media containing kanamycin and grown at 37°C for approximately 4 hours. When the cells reached log phase growth, IPTG was added to a final concentration of 1 mM and arabinose to a final concentration of 0.2% were added to the ''E. coli'' bacterial culture to induce production of TDP43-RRM12. The protein induction occurred for 2.5 hr at 37°C then the cells were collected by centrifugation at 3000 g for 10 min. The harvested cells were snap frozen at -80 °C. | ||
| + | |||
| + | #Retrieve your BL21-A1 pET_MBP_SNAP_TDP43-RRM12 cell pellet from the front laboratory bench and leave it on your bench to thaw. | ||
| + | #Obtain a 3 mL aliquot of lysis buffer from the front laboratory bench. | ||
| + | #*Calculate volume of imidazole needed in the lysis buffer such that the final concentration of imidazole is 10 mM (stock concentration = 2.5 M). | ||
| + | #Resuspend each pellet completely in 1.5 mL of the lysis buffer with imidazole. | ||
| + | # Add 5μL of the lysonase to each cell suspension. | ||
| + | #Incubate at room temperature for 10 min on a nutator. | ||
| + | #A sonicator will be used to lyse the cells. In a process called soniporation, sound energy is produced via a sonicator and exerted through a probe that rests in the sample liquid. This results in lysis of the cell membrane which releases the cell contents. The Instructor will lyse your sample with the sonicator in the 4 °C cold room using the following settings: 50% duty cycle for 4 min at power 4. | ||
| + | #*Because sonication is more effective when completed with larger volumes, you will combine your samples with those of two other teams. The Instructor will inform you which team samples should be combined. | ||
| + | #Divide cell lysate evenly amongst the teams combined for sonication (~1500 μL per tube) and centrifuge at 12,500g for 30 min. | ||
| + | |||
| + | ===Part 3: Prepare Ni-NTA affinity column=== | ||
| + | #Obtain a 500 μL aliquot of 50% slurry (Ni-NTA resin) from the front laboratory bench, mix the slurry by inverting the tube several times. | ||
| + | #Centrifuge the slurry for 30 sec then remove the supernatent. | ||
| + | #To wash the slurry, add 500 μL of 1X PBS and invert the tube 3 times. | ||
| + | #Add the slurry to the column and allow the 1X PBS to run through the column. | ||
| + | #*Be sure a beaker is placed under the column to collect the waste! | ||
| + | #When the PBS has flowed through, cap the bottom and the top of the column until you are ready to add the cell lysate in Part #4. | ||
| + | |||
| + | ===Part 4: Purify TDP43-RRM12=== | ||
| + | #Retrieve your cell pellets from the centrifuge and transfer the supernatent to a fresh 2 mL tube. | ||
| + | #*Label the tube containing the cell pellet and give it to the teaching faculty! | ||
| + | #*'''Note: It is VERY important that you collect fraction samples as indicated in Step 2, Step 8, & Step 9!''' | ||
| + | #Transfer 30 μL into labeled 1.5 mL eppendorf tubes and give the aliquots to the teaching faculty. Also label and save the pellet at this step. | ||
| + | #*You will use this aliquot to examine protein yield prior to purification using polyacrylamide gel electrophoresis (PAGE) on a later date. At that time, the teaching faculty will provide you with a bacterial pellet from uninduced bacteria to compare. | ||
| + | #*'''Note:''' Why would you want to compare induced to uninduced bacteria? | ||
| + | #Add 10 μL of the SnapTag substrate to the supernatant. | ||
| + | #Next add DTT to the supernatant at a final concentration of 1 mM. | ||
| + | #*'''Note:''' the SnapTag substrate / buffer reaction allows the TDP43 protein to be labeled with a 647nm fluorophore at the SNAP sequence incorporated into the protein construct. | ||
| + | #Transfer the supernatant to the slurry prepared in Part 3. | ||
| + | #*'''Note:''' the slurry consists of a nickel resin that will bind His-tagged proteins. | ||
| + | #Use aluminum foil to wrap the tube such that the supernatent / slurry are protected from light. | ||
| + | #Incubate the supernatent / slurry for 2 hr (or until 4:30p, be sure to record the exact incubation time in your notebook!) in the 4 °C cooler on a nutator. | ||
| + | #*What is happening during this incubation? Hint: consider the purpose of the SnapTag and that of the nickel resin slurry. | ||
| + | #Following the incubation, reclamp the column in the ring stand to collect the flowthrough. | ||
| + | #*Prepare a 1.5 mL tube under the column to collect the flow through (liquid that drips from the column). Only remove the bottom cap when you are ready to collect all flow through. | ||
| + | #To wash the slurry in the column, add 2.5 mL of 1X PBS containing 10 mM imidazole. | ||
| + | #*Collect ~250 μL of the first wash flowthrough (in a well-labeled 1.5 mL tube), the rest of the flowthrough is waste and will be collected in the beaker under the column. | ||
| + | #*What is in the flowthrough? What is attached to the nickel resin slurry? | ||
| + | #Repeat the 2.5 mL of 1X PBS with imidazole wash step a total of 3 times. | ||
| + | #Next, wash the slurry with 2 mL of HRV 3C buffer. | ||
| + | #Cap the bottom of the column. | ||
| + | #*'''Note:''' It is very important that the column is capped before the next step, if not the protein will be lost in the waste beaker! | ||
| + | #In the remaining 1 mL of HRV 3C buffer, add 10 μL of HRV 3C protease. Then add the HRV 3C buffer / protease to the capped column. | ||
| + | #Resuspend the slurry in the HRV 3C buffer / protease by pipetting up and down with a P1000. | ||
| + | #Add a cap to the top of the column and wrap the column in aluminum foil. | ||
| + | #Incubate the slurry overnight at 4 °C on a rotator wheel. | ||
| + | #The next steps will be completed by the teaching faculty, but should be included in your methods! | ||
| + | #*Following the overnight incubation, uncap the base of the column and collect all flow through. | ||
| + | #*Resuspend the slurry in 250 μL 1X PBS and transfer to a fresh tube. | ||
| + | |||
| + | ==Reagents== | ||
| + | For gel electrophoresis: | ||
| + | *1% agarose in water, with 10 uL SYBR Safe DNA gel stain per 100 uL agarose solution | ||
| + | |||
| + | *1X TAE gel electrophoresis buffer: (BioRad) | ||
| + | **40 mM Tris | ||
| + | **20 mM acetic acid | ||
| + | **1 mM EDTA | ||
| + | |||
| + | *6X gel loading dye, blue (NEB) | ||
| + | |||
| + | *1 kb DNA ladder (NEB) | ||
| + | |||
| + | For protein induction / purification: | ||
| + | |||
| + | *LB (Luria-Bertani broth, BD Biosciences) | ||
| + | **1% Tryptone | ||
| + | **0.5% Yeast Extract | ||
| + | **1% NaCl | ||
| + | ** autoclaved for sterility | ||
| + | *Isopropyl β-d-1-thiogalactopyranoside (IPTG), stock 100mM | ||
| + | *Arabinose (Sigma), stock 20% | ||
| + | *Kanamycin (Sigma): 50 mg/mL, 1000X stock | ||
| + | *PBS: phosphate saline buffer (VWR) | ||
| + | *Ni-NTA agarose (Qiagen) | ||
| + | *Lysis buffer | ||
| + | **50mM NaH2PO4 | ||
| + | **300mM NaCl | ||
| + | **10mM imidazole | ||
| + | **Lysonase (VWR) | ||
| + | *Imidazole (Sigma), stock 2.5M | ||
| + | *SNAP-Surface® Alexa Fluor® 647 (NEB) | ||
| + | *DTT : DL-dithiothreitol (Sigma), stock 1M | ||
| + | *HRV 3C Protease (ThermoFisher) | ||
| + | |||
| + | ==Navigation links== | ||
| + | Next day: [[20.109(F20):M2D3 | Assess purity and concentration of purified protein]]<br> | ||
| + | Previous day: [[20.109(F20):M2D1 | Complete in silico cloning of protein expression plasmid]] | ||
Revision as of 00:52, 17 July 2020

Contents
Introduction
To induce production of PF3D7_1351100 protein from the expression plasmid that was 'cloned' in the previous laboratory session, a lactose-analogue isopropyl β-D-1-thiogalactopyranoside (IPTG) and arabinose was used to induce expression in Nico(DE3) E. coli bacterial cells. The use of IPTG to induce protein expression is based on the native lac operon used for lactose metabolism in bacterial cells.

The native lac operon is a powerful tool in engineering protein expression systems because it enables researchers to control gene expression using inducer molecules. The lacZYA genes are only expressed when lactose is present. If a gene of interest is cloned downstream of the operator sequence, the expression of this gene can be controlled by LacI repression and lactose derepression. To further control the system for protein expression, IPTG is used as a lactose-analog as it is not metabolized by the cells.

Today you will isolate the expressed PF3D7_1351100 protein from the bacterial cells. Remember that the PF3D7_1351100 gene sequence was synthesized using a gBlock and 6xHis-tag was added to the DNA sequence. The resultant protein is therefore His-tagged. Histidine has several interesting properties, notably its near-neutral pKa, and His-rich peptides are promiscuous binders, particularly to metals. (For example, histidine side chains help coordinate iron molecules in hemoglobin.)
You will use a nickel-agarose resin to separate PF3D7_1351100 from the other proteins present in the bacteria. The His-tagged protein will preferentially bind to the nickel-coated beads, while proteins irrelevant to our purposes can be washed away using imidazole. Remember, the Nico(DE3) cells are not only producing the PF3D7_1351100 protein, but also the proteins needed for cellular function and survival. Because imidazole is also able to bind to nickel, washing the column with a low concentration solution will promote competition for binding between the imidazole and bound proteins. Proteins that are non-specifically bound will have a lower affinity for the nickel than imidazole and be washed from the column, whereas the 6xHis-tagged PF3D7_1351100 will remain adhered to the nickel-agarose resin.

Protocols
Part 1: Electrophorese confirmation digests
Electrophoresis is a technique that separates large molecules by size using an applied electrical field and a sieving matrix. DNA, RNA and proteins are the molecules most often studied with this technique; agarose and acrylamide gels are the two most common sieves. The molecules to be separated enter the matrix through a well at one end and are pulled through the matrix when a current is applied across it. The larger molecules get entwined in the matrix and are stalled; the smaller molecules wind through the matrix more easily and travel farther away from the well. The distance a DNA fragment travels is inversely proportional to the log of its length. Over time fragments of similar length accumulate into “bands” in the gel. Higher concentrations of agarose can be used to resolve smaller DNA fragments.

DNA and RNA are negatively charged molecules due to their phosphate backbone, and they naturally travel toward the positive electrode at the far end of the gel. Today you will separate DNA fragments using an agarose matrix. Agarose is a polymer that comes from seaweed. To prepare these gels, agarose and 1X TAE buffer (Tris base, acetic acid, and EDTA) are microwaved until the agarose is melted and fully dissolved. The molten agar is then poured into a horizontal casting tray, and a comb is added. Once the agar has solidified, the comb is removed, leaving wells into which the DNA samples can be loaded.
You will use a 1% agarose gel with SYBR Safe DNA stain (prepared by the teaching faculty) to separate the DNA fragments in your four digested samples as well as a reference lane of molecular weight markers (also called a DNA ladder).
- Add 5 μL of 6x loading dye to the digests.
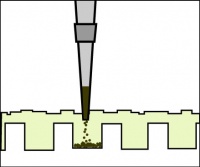 Illustration of proper gel loading technique.
Illustration of proper gel loading technique.
- Loading dye contains bromophenol blue as a tracking dye to follow the progress of the electrophoresis (so you don’t run the smallest fragments off the end of your gel!) as well as glycerol to help the samples sink into the wells.
- Flick the eppendorf tubes to mix the contents, then quick spin them in the microfuge to bring the contents of the tubes to the bottom.
- Load 25 μL of each digest into the gel, as well as 10 μL of 1kb DNA ladder.
- Be sure to record the order in which you load your samples!
- To load your samples, draw the volume listed above into the tip of your P200 or P20. Lower the tip below the surface of the buffer and directly over the well. Avoid lowering the tip too far into the well itself so as to not puncture the well. Expel your sample slowly into the well. Do not release the pipet plunger until after you have removed the tip from the gel box (or you'll draw your sample back into the tip!).
- Once all the samples have been loaded, attach the gel box to the power supply and electrophorese the gel at 125 V for 45 minutes.

Part 2: Lyse BL21-A1 pET_MBP_SNAP_TDP43-RRM12 cells
Previously, the teaching faculty inoculated 5 mL of LB media containing kanamycin with BL21-A1 pET_MBP_SNAP_TDP43-RRM12, incubated the culture at 37 °C overnight. The next day the overnight bacterial culture was diluted 1:10 in 50 mL total of fresh LB media containing kanamycin and grown at 37°C for approximately 4 hours. When the cells reached log phase growth, IPTG was added to a final concentration of 1 mM and arabinose to a final concentration of 0.2% were added to the E. coli bacterial culture to induce production of TDP43-RRM12. The protein induction occurred for 2.5 hr at 37°C then the cells were collected by centrifugation at 3000 g for 10 min. The harvested cells were snap frozen at -80 °C.
- Retrieve your BL21-A1 pET_MBP_SNAP_TDP43-RRM12 cell pellet from the front laboratory bench and leave it on your bench to thaw.
- Obtain a 3 mL aliquot of lysis buffer from the front laboratory bench.
- Calculate volume of imidazole needed in the lysis buffer such that the final concentration of imidazole is 10 mM (stock concentration = 2.5 M).
- Resuspend each pellet completely in 1.5 mL of the lysis buffer with imidazole.
- Add 5μL of the lysonase to each cell suspension.
- Incubate at room temperature for 10 min on a nutator.
- A sonicator will be used to lyse the cells. In a process called soniporation, sound energy is produced via a sonicator and exerted through a probe that rests in the sample liquid. This results in lysis of the cell membrane which releases the cell contents. The Instructor will lyse your sample with the sonicator in the 4 °C cold room using the following settings: 50% duty cycle for 4 min at power 4.
- Because sonication is more effective when completed with larger volumes, you will combine your samples with those of two other teams. The Instructor will inform you which team samples should be combined.
- Divide cell lysate evenly amongst the teams combined for sonication (~1500 μL per tube) and centrifuge at 12,500g for 30 min.
Part 3: Prepare Ni-NTA affinity column
- Obtain a 500 μL aliquot of 50% slurry (Ni-NTA resin) from the front laboratory bench, mix the slurry by inverting the tube several times.
- Centrifuge the slurry for 30 sec then remove the supernatent.
- To wash the slurry, add 500 μL of 1X PBS and invert the tube 3 times.
- Add the slurry to the column and allow the 1X PBS to run through the column.
- Be sure a beaker is placed under the column to collect the waste!
- When the PBS has flowed through, cap the bottom and the top of the column until you are ready to add the cell lysate in Part #4.
Part 4: Purify TDP43-RRM12
- Retrieve your cell pellets from the centrifuge and transfer the supernatent to a fresh 2 mL tube.
- Label the tube containing the cell pellet and give it to the teaching faculty!
- Note: It is VERY important that you collect fraction samples as indicated in Step 2, Step 8, & Step 9!
- Transfer 30 μL into labeled 1.5 mL eppendorf tubes and give the aliquots to the teaching faculty. Also label and save the pellet at this step.
- You will use this aliquot to examine protein yield prior to purification using polyacrylamide gel electrophoresis (PAGE) on a later date. At that time, the teaching faculty will provide you with a bacterial pellet from uninduced bacteria to compare.
- Note: Why would you want to compare induced to uninduced bacteria?
- Add 10 μL of the SnapTag substrate to the supernatant.
- Next add DTT to the supernatant at a final concentration of 1 mM.
- Note: the SnapTag substrate / buffer reaction allows the TDP43 protein to be labeled with a 647nm fluorophore at the SNAP sequence incorporated into the protein construct.
- Transfer the supernatant to the slurry prepared in Part 3.
- Note: the slurry consists of a nickel resin that will bind His-tagged proteins.
- Use aluminum foil to wrap the tube such that the supernatent / slurry are protected from light.
- Incubate the supernatent / slurry for 2 hr (or until 4:30p, be sure to record the exact incubation time in your notebook!) in the 4 °C cooler on a nutator.
- What is happening during this incubation? Hint: consider the purpose of the SnapTag and that of the nickel resin slurry.
- Following the incubation, reclamp the column in the ring stand to collect the flowthrough.
- Prepare a 1.5 mL tube under the column to collect the flow through (liquid that drips from the column). Only remove the bottom cap when you are ready to collect all flow through.
- To wash the slurry in the column, add 2.5 mL of 1X PBS containing 10 mM imidazole.
- Collect ~250 μL of the first wash flowthrough (in a well-labeled 1.5 mL tube), the rest of the flowthrough is waste and will be collected in the beaker under the column.
- What is in the flowthrough? What is attached to the nickel resin slurry?
- Repeat the 2.5 mL of 1X PBS with imidazole wash step a total of 3 times.
- Next, wash the slurry with 2 mL of HRV 3C buffer.
- Cap the bottom of the column.
- Note: It is very important that the column is capped before the next step, if not the protein will be lost in the waste beaker!
- In the remaining 1 mL of HRV 3C buffer, add 10 μL of HRV 3C protease. Then add the HRV 3C buffer / protease to the capped column.
- Resuspend the slurry in the HRV 3C buffer / protease by pipetting up and down with a P1000.
- Add a cap to the top of the column and wrap the column in aluminum foil.
- Incubate the slurry overnight at 4 °C on a rotator wheel.
- The next steps will be completed by the teaching faculty, but should be included in your methods!
- Following the overnight incubation, uncap the base of the column and collect all flow through.
- Resuspend the slurry in 250 μL 1X PBS and transfer to a fresh tube.
Reagents
For gel electrophoresis:
- 1% agarose in water, with 10 uL SYBR Safe DNA gel stain per 100 uL agarose solution
- 1X TAE gel electrophoresis buffer: (BioRad)
- 40 mM Tris
- 20 mM acetic acid
- 1 mM EDTA
- 6X gel loading dye, blue (NEB)
- 1 kb DNA ladder (NEB)
For protein induction / purification:
- LB (Luria-Bertani broth, BD Biosciences)
- 1% Tryptone
- 0.5% Yeast Extract
- 1% NaCl
- autoclaved for sterility
- Isopropyl β-d-1-thiogalactopyranoside (IPTG), stock 100mM
- Arabinose (Sigma), stock 20%
- Kanamycin (Sigma): 50 mg/mL, 1000X stock
- PBS: phosphate saline buffer (VWR)
- Ni-NTA agarose (Qiagen)
- Lysis buffer
- 50mM NaH2PO4
- 300mM NaCl
- 10mM imidazole
- Lysonase (VWR)
- Imidazole (Sigma), stock 2.5M
- SNAP-Surface® Alexa Fluor® 647 (NEB)
- DTT : DL-dithiothreitol (Sigma), stock 1M
- HRV 3C Protease (ThermoFisher)
Next day: Assess purity and concentration of purified protein