20.109(F20):M2D1

Contents
- 1 Introduction
- 2 Protocols
- 2.1 Part 1: Synthesis and restriction enzyme digest of PF3D7_1351100 insert
- 2.2 Part 2: Restriction enzyme digest of pET-28b(+) expression vector
- 2.3 Part 3: Ligation of PF3D7_1351100 insert and pET-28b(+) expression vector
- 2.4 Part 4: Confirmation digest of pET-28b(+)_PF3D7_1351100
- 2.5 Part 5: Electrophorese confirmation digests
- 3 Reagents list
- 4 Navigation links
Introduction
Though the theme of Module 2 is focused on screening for small molecules that bind the PF3D7_1351100 protein, today will focus on a few key techniques used in DNA engineering. Because the sequence of proteins is determined by the sequence of the genes that encode them, learning how to manipulate DNA is an important first step. Today you will complete the cloning steps used to incorporate the gene that encodes the PF3D7_1351100 protein into an expression vector. The expression vector contains the genetic elements needed to express and purify a protein of interest.
Expression vectors contain several features important for cloning, plasmid replication, and protein expression -- all of which are important for purifying high-quality protein. To generate this expression plasmid, two common DNA engineering techniques were used: restriction enzyme digestion and ligation. First, the PF3D7 insert was synthesized such that the gene sequence is flanked by restriction enzymes sites. Next, this fragment and the vector were digested to create compatible ends. Last, the compatible ends of the digested insert and vector were ligated to generate the pET-28b(+)_PF3D7_1351100 expression plasmid.

Restriction enzyme digest

Restriction endonucleases, also called restriction enzymes, 'cut' or 'digest' DNA at specific sequences of bases. The restriction enzymes are named according to the prokaryotic organism from which they were isolated. For example, the restriction endonuclease EcoRI (pronounced “echo-are-one”) was originally isolated from E. coli giving it the “Eco” part of the name. “RI” indicates the particular version on the E. coli strain (RY13) and the fact that it was the first restriction enzyme isolated from this strain.
The sequence of DNA that is bound and cleaved by an endonuclease is called the recognition sequence or restriction site. These sequences are usually four or six base pairs long and palindromic, that is, they read the same 5’ to 3’ on the top and bottom strand of DNA. For example, the recognition sequence for EcoRI is 5’ GAATTC 3’ (see figure at right). EcoRI cleaves the phosphate backbone of DNA between the G and A of the recognition sequence, which generates overhangs or 'sticky ends' of double-stranded DNA.
Unlike EcoRI, some other restriction enzymes cut precisely in the middle of the palindromic DNA sequence, thus leaving no overhangs after digestion. The single-stranded overhangs resulting from DNA digestion by enzymes such as EcoRI are called sticky ends, while double-stranded ends resulting from digestion by enzymes such as HaeIII are called blunt ends. HaeIII recognizes 5’ GGCC 3’ and upon recognition cuts in the center of the sequence.
Ligation

In a ligation reaction, DNA ends are covalently attached to one another via the ligase enzyme. The efficiency of the reaction is related to type of DNA ends: compatible sticky ends will ligate more efficiently than blunt ends, and non-compatible sticky ends will not be ligated due to the lack of hydrogen bonding between the basepairs. To initiate the ligation reaction, hydrogen bonds are formed between the compatible overhangs of DNA fragments. The ligase enzyme then forms a covalent phosphodiester bond between the 3' hydroxyl end of the 'acceptor' nucleotide and the 5' phosphodiester end of the 'donor' nucleotide.
The first step in this process is the addition of AMP (adenylation) to a lysine residue within the active site of DNA ligase, which releases a pyrophosphate. Next, the AMP is transferred to the 5' phosphate of the donor nucleotide resulting in the formation of a pyrophosphate bond. Lastly, a phosphodiester bond is formed between the 5' phosphate of the donor nucleotide and the 3' hydroxyl of the 3' acceptor nucleotide.
Protocols
Because DNA engineering at the benchtop can take days, if not weeks, you will clone the expression plasmid in silico today. You can use any DNA manipulation software you choose to complete the protocols, but the instructions provided are for SnapGene. Please note that if you use a different program the Instructors may not be able to assist you.
Part 1: Synthesis and restriction enzyme digest of PF3D7_1351100 insert
To use SnapGene software off campus you must log into a VPN connection prior to opening the SnapGene. Here is the link to the VPN download and installation instructions. Also you will need to update the SnapGene license number if you have not opened the application since March. The new license information can be found here.
As discussed in the M2 project overview, PF3D7_1351100 is an essential protein of unknown function in Plasmodium falciparum. In an effort to study PF3D7_1351100 a researcher in the Niles Laboratory, Dr. Khan Osman, cloned the gene that encodes the protein into an expression plasmid. Rather than amplifying the gene from the P. falciparum genome, gBlock synthesis technology was used. A gBlock is a double-stranded DNA segment that is synthesized commercially without the use of live cells. To generate a gBlock, a researcher simply submits the basepair sequence to be synthesized. This method is useful for several reasons including: 1. it can be technically difficult to amplify genes from certain organisms, and 2. it can be easier to modify DNA that is synthetically generated. For this project, the PF3D7_1351100 gBlock sequence was modified such that the codon usage was optimized for expression in E. coli cells.

- Open the word document with the PF3D7_1351100 insert sequence (linked here).
- Open SnapGene. From the options, select 'New DNA File...'.
- Copy and paste the sequence from the .docx file above.
- Enter "PF3D7_1351100" for the File Name (in the lower, right corner), select 'linear' for the topology (in the lower, left corner), then click 'OK'.
- A new window will open with a map of PF3D7_1351100 showing the unique restriction enzyme sites within the sequence.
- In later steps you will generate a map of the PF3D7_1351100 insert cloned into the pET-28b(+) expression vector. To make the map more visually useful, create a feature that defines the PF3D7_1351100 insert.
- Click 'Sequence' from the options at the bottom of the window.
- Highlight the entire sequence in the window.
- From the toolbar, select 'Features' → 'Add Feature...'
- In the new window name, type PF3D7_1351100 into the 'Feature:' box.
- Select gene from the dropdown in the 'Type:' box and select the right facing arrowhead (this denotes the directionality of the insert).
- Then click 'OK'.
- The PF3D7_1351100 gBlock was modified such that a 6xHis-tag sequence was added to the N-terminal end of the protein. 6xHis-tags are added for protein purification.
- In your laboratory notebook, draw a schematic diagram that shows the following:
- The gene sequence (as a line) with 5' and 3' orientation denoted.
- The associated protein sequence (again, as a line) with the N' and C' termini denoted.
- The location of the 6xHis-tag on the gene sequence and protein sequence.
- Add the 6xHis-tag (5' MGSSHHHHHHSSG 3') to the PF3D7_1351100 insert sequence by setting the cursor to the location in the DNA sequence. Then begin typing the 6xHis-tag sequence. A new window will appear with the typed bases. Confirm the bases are entered correctly, then click 'Insert'.
- Use the steps above to define the 6xHis-tag sequence as a feature.
- As shown in the schematic of our cloning strategy, NcoI and BamHI recognition sequences were added to the PF3D7_1351100 gBlock to enable cloning into the pET-28b(+) vector. Specifically, NcoI was added to the 5' end and BamHI to the 3' end of the PF3D7_1351100 sequence.
- Search the NEB enzyme list to find the NcoI and BamHI recognition sequences.
- In your laboratory notebook, complete the following:
- Record the recognition sequences for NcoI and BamHI. Include the cleavage locations within each sequence.
- Include the recognition sites for NcoI and BamHI to the schematic diagram created above. Should these recognition sites be included on the gene sequence or the protein sequence?
- Add the NcoI and BamHI recognition sites to the PF3D7_1351100 insert sequence by setting the cursor to the location in the DNA sequence, then begin typing. A new window will appear with the typed bases. Confirm the bases are entered correctly, then click 'Insert'.
- Now that you have the PF3D7_1351100 gBlock, you need to digest with NcoI and BamHI to generate 'sticky ends' that will enable you to ligate the PF3D7_1351100 insert into the pET-28b(+) vector.
- On the map of the PF3D7_1351100 insert, select the NcoI recognition site by clicking on the enzyme name. Then hold the shift key and select the BamHI recognition site.
- This should highlight the area between the enzyme recognition sites.
- Click the drop-down arrow next to the 'Copy' icon at the top of the window.
- Select 'Copy Restriction Fragment.'
- Click the drop-down arrow next to the 'New' icon at the top of the window.
- Select 'New DNA File...'.
- Paste the restriction fragment from the previous step in the text box, then click 'OK'.
- A new window will open with the digested PF3D7_1351100 insert.
- In your laboratory notebook, complete the following:
- Record the length of the insert. How does the length of the insert compare to the length of the gBlock.
- Is the insert double-stranded or single-stranded?
- Is the insert a blunt end product or sticky end product?
- Save the insert file.
Part 2: Restriction enzyme digest of pET-28b(+) expression vector
For the ligation step, it is important to generate compatible 'sticky ends' on the insert and vector. Above, you digested your PF3D7_1351100 insert with NcoI and BamHI in a double-digest to prepare the insert for your cloning. Here you will digest the pET-28b(+) vector to create compatible ends that can be ligated.

- Open the word document with the pET-28b(+) vector sequence (linked here).
- Copy and paste the vector sequence into a New DNA File window and save this sequence.
- Be sure to select circular from the topology options.
- One very useful aspect of SnapGene is that the software is able to recognize features, or sequences that match known genes and binding sites, in DNA sequences. A window titled "Detect Common Features" should appear.
- In your laboratory notebook, include a summary of the details provided about features in the pET-28b(+) vector.
- Select 'Add Features'.
- A new window will open with a map of the vector showing the unique restriction enzyme sites and annotated features within the sequence.
- To generate the sticky ends that will enable you to ligate the PD3D7_1351100 insert into the vector, view the map of your vector sequence.
- Select the NcoI recognition site by clicking on the enzyme name, then hold the shift key and select the BamHI recognition site.
- Select 'Actions' --> 'Restriction and Insertion Cloning' --> 'Delete Restriction Fragment...' from the toolbar.
- In your laboratory notebook, complete the following:
- What is the length of the digested vector product?
- How many basepairs were removed (compared to the intact cloning vector)?
Part 3: Ligation of PF3D7_1351100 insert and pET-28b(+) expression vector
Before you prepare a ligation, one very important step is to calculate the amounts of DNA that will be used in the reaction. Ideally, you should use a 3:1 molar ratio of insert to vector (note: it is a molar ratio, not a volumetric ratio!). You will use the steps below to calculate the volume amount (based on the molar ratio!) of the PF3D7_1351100 insert and pET-28b(+) expression vector you would use to complete this ligation in the laboratory.

Use the following information to calculate the volume of insert and vector needed to prepare a ligation with a 3:1 molar ratio (insert:vector).
- Concentration of PF3D7_1351100 insert solution = 25 ng/uL
- Concentration of pET-28b(+) expression vector solution = 50 ng/uL
- Molecular weight of a basepair = 660 g/mol
- Sizes, in basepairs, of the insert and vector sequences (this was determined in the exercises above!)
Though there are are different strategies that can be used to complete the ligation calculations, it may be easier to break the math into the following steps:
- Determine the volume of vector that will be used in the ligation reaction.
- Typically, it is best to use 50 - 100 ng of vector.
- Calculate the moles of vector.
- Calculate the moles of insert.
- Remember, this number should be 3-fold more than the moles of vector to accomplish a 3:1 molar ratio.
- Calculate the volume of insert that contains the appropriate moles of insert.
- One additional consideration is the volume of the reaction. The total volume of the ligation reaction should not be greater than 15 μL. In this, the total volume of the insert and vector should not be greater than 13.5 μL as additional reagents are required in the reaction.
- If the insert and vector volume total greater than 13.5 μL, you should (1) scale down both DNA amounts, using less than 50 ng backbone and/or (2) stray from the ideal 3:1 molar ratio.
- You may ask the teaching faculty for advice during class if you are unsure what choice is best.
- In your laboratory notebook, calculate the volume of insert and volume of vector that should be used for a ligation reaction that contains a 3:1 molar ratio of insert:vector. Show all math!
- Feel free to take a picture of your hand-written work and embed the image in your notebook.
- Next you will complete this ligation in silico to generate a map, or visual representation, of the pET-28b(+)_PF3D7_1351100 plasmid.
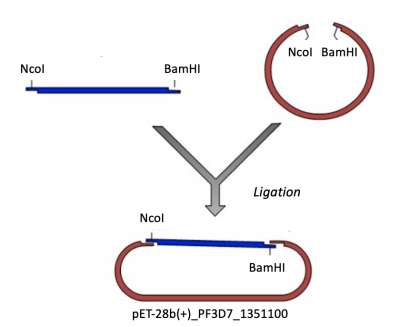

- To ligate the PF3D7_1351100 insert into the pET-28b(+) expression vector, select 'Actions' --> 'Restriction and Insertion Cloning' --> 'Insert Fragment...'.
- A new window will open. In the bottom workspace of the window, a cloning schematic will appear showing a vector and insert icon.
- Click on the 'Vector' label. Then in the workspace at the the right of the window, select the vector file from the 'Vector:' drop-down.
- Select the restriction enzymes used to digest the expression vector from the drop-down boxes next to the text boxes that contain 'cut'.
- Next, click on the 'Insert' label at the bottom of the window and complete the steps as done for the expression vector.
- For the insert, use the PF3D7_1351100 undigested file.
- Click 'Clone'.
- A new window will open with the cloned pET-28b(+)_PF3D7_1351100 product!
- In your laboratory notebook, complete the following:
- What is the size of the plasmid? Does this make sense given the lengths of the insert and vector?
- Does your sequence still contain a NcoI recognition sequence? A BamHI recognition sequence?

Part 4: Confirmation digest of pET-28b(+)_PF3D7_1351100
To confirm the pET-28b(+)_PF3D7_1351100 construct that we will use for this module, you will perform a 'diagnostic' or 'confirmation' digest. As discussed in prelab, this step is an important control -- you want to be sure that the products you use in your research are correct! This step is used to check products you clone yourself and, perhaps more importantly, those that you may receive from another researcher.
Ideally you will use a single enzyme that cuts once within the vector and once within your insert. Unfortunately, this is rarely an option and you instead need to select an enzyme that cuts once within the vector and a second, compatible enzyme that cuts once within the insert. Enzyme compatibility is determined by the buffer. If two enzymes are active, or able to cleave DNA, in the same buffer, they are compatible. The NEB double digest online tool will prove very helpful in identifying compatible enzyme combinations!
Use the information from prelab, the 20.109 list of enzymes (linked here), and the plasmid map you generated above to choose the enzymes you will use.
- To choose restriction enzymes for your confirmation digest, look at the plasmid map for your pET-28b(+)_PF3D7_1351100 construct.
- Identify possible sites that will enable to you confirm the pET-28b(+)_PF3D7_1351100 sequence.
- Remember the guidelines discussed in prelab!
- After you identify the enzymes that you will use for the confirmation digest, complete a virtual digest in using the pET-28b(+)_PF3D7_1351100 map you generated above.
- On the map of pET-28b(+)_PF3D7_1351100, select the first recognition site by clicking on the enzyme name. Then hold the shift key and select the second recognition site.
- Select 'Tools' --> 'Simulate Agarose Gel' from the toolbar.
- In your laboratory notebook, complete the following:
- Record the expected fragment sizes from the confirmation digest.
- Are the fragments distinct or ambiguously close together?
- Now that you identified which enzyme(s) to use in your confirmation digest, consider which controls should be included to ensure the results are interpretable.
- In your laboratory notebook, explain why the following reactions are included as controls for the confirmation digest experiment:
- Undigested pET-28b(+)_PF3D7_1351100.
- Single digests of pET-28b(+)_PF3D7_1351100 (each enzyme used alone in a digest with pET-28b(+)_PF3D7_1351100).
- Use the table below to calculate the volumes of each reagent that should be included in the confirmation digest reactions.
Keep the following in mind as you consider which enzymes to use:
- Each enzyme should be present in 10 U quantity per reaction. As an example, the XbaI vial contains 20,000 U/mL, or 20 U/μL.
- The 20.109 enzyme stocks are always the "S" size and concentration when you search for them on the NEB website.
- Enzyme volume should not exceed 10% of the total reaction volume to prevent star activity due to excess glycerol.
- To find the concentration of the enzyme(s) you choose, search the NEB site.
| Diagnostic digest (enzyme #1 AND enzyme #2) |
Enzyme #1 ONLY | Enzyme #2 ONLY | Uncut (NO enzyme) | |
|---|---|---|---|---|
| pET-28b(+)_PF3D7_1351100 | 5 μL | 5 μL | 5 μL | 5 μL |
| 10X NEB buffer (buffer name:____________) |
2.5 μL | 2.5 μL | 2.5 μL | 2.5 μL |
| Enzyme #1 (enzyme name:____________) |
____ μL | ____ μL | ||
| Enzyme #2 (enzyme name:____________) |
____ μL | ____ μL | ||
| H2O | to a final volume of 25 μL | |||
To ensure the steps required for preparing a digest are clear, the Instructor will provide a live demonstration of this process. You should provide a written description of the procedure in your laboratory notebook!
In your laboratory notebook, complete the following:
- Provide a written overview / description of the the procedure used to prepare a restriction enzyme digest (from the live demonstration).
- For how long will the digests incubate and at what temperature?
Part 5: Electrophorese confirmation digests
Electrophoresis is a technique that separates large molecules by size using an applied electrical field and a sieving matrix. DNA, RNA and proteins are the molecules most often studied with this technique; agarose and acrylamide gels are the two most common sieves. The molecules to be separated enter the matrix through a well at one end and are pulled through the matrix when a current is applied across it. The larger molecules get entwined in the matrix and are stalled; the smaller molecules wind through the matrix more easily and travel farther away from the well. The distance a DNA fragment travels is inversely proportional to the log of its length. Over time fragments of similar length accumulate into “bands” in the gel. Higher concentrations of agarose can be used to resolve smaller DNA fragments.

DNA and RNA are negatively charged molecules due to their phosphate backbone, and they naturally travel toward the positive electrode at the far end of the gel. Today you will separate DNA fragments using an agarose matrix. Agarose is a polymer that comes from seaweed. To prepare these gels, agarose and 1X TAE buffer (Tris base, acetic acid, and EDTA) are microwaved until the agarose is melted and fully dissolved. The molten agar is then poured into a horizontal casting tray, and a comb is added. Once the agar has solidified, the comb is removed, leaving wells into which the DNA samples can be loaded.
For the digests that were prepared in the previous laboratory session, a 1% agarose gel with SYBR Safe DNA stain was used to separate the DNA fragments in the four digest reactions. In addition, a well was loaded with a molecular weight marker (also called a DNA ladder) to determine the size of the fragments.
To ensure the steps included below are clear, please watch the video tutorial linked here: [DNA gel electrophoresis]. The steps are detailed below so you can follow along!
- Add 5 μL of 6x loading dye to the digests.
- Loading dye contains bromophenol blue as a tracking dye, which enables you to follow the progress of the electrophoresis.
- Glycerol is also included to weight the samples such that the liquid sinks into well.
- Flick the eppendorf tubes to mix the contents, then quick spin them in the microfuge to bring the contents of the tubes to the bottom.
- Load 25 μL of each digest into the gel, as well as 10 μL of 1kb DNA ladder.
- Be sure to record the order in which you load your samples!
- To load your samples, draw the volume listed above into the tip of your P200 or P20. Lower the tip below the surface of the buffer and directly over the well. Avoid lowering the tip too far into the well itself so as to not puncture the well. Expel your sample slowly into the well. Do not release the pipet plunger until after you have removed the tip from the gel box (or you'll draw your sample back into the tip!).
- Once all the samples have been loaded, attach the gel box to the power supply and electrophorese the gel at 125 V for 45 minutes.
- Lastly, visualize the DNA fragments in the agarose gel using the gel documentation system.
Reagents list
- pET-28b(+)_PF3D7_1351100 (concentration = 25 ng/μL) (a gift from the Niles Laboratory)
- 10X buffer; the buffer will depend on the enzymes you use for your confirmation digest (from NEB)
- restriction enzyme(s); the concentration of each enzyme is listed on the product information page (from NEB)
- 1% agarose in 1X TAE (agarose from VWR)
- with 10% (v/v) μL SYBR Safe DNA stain (from Invitrogen)
- 1X TAE gel electrophoresis buffer: 40 mm Tris, 20 mM acetic acid, 1 mM EDTA (from BioRad)
- 6X gel loading dye, blue (from NEB)
- 1 kb DNA ladder (from NEB)
Next day: Perform protein purification protocol