20.109(S22):M2D7

Contents
Introduction

The core RNAP enzyme is composed of five subunits: β, β', αI, αII, and ω. The β and β' subunits catalyze the formation of phospodiester bonds in elongating transcripts and form a 'claw' structure that is joined together by the α-dimer. The N-terminal domains of the α-dimer are in contact with the β and β' subunits and the C-terminal domains contain a flexible linker that is able to associate with DNA and / or regulatory proteins. The ω subunit is involved in assembly of RNAP. When this core enzyme is complexed with a σ factor, the holoenzyme is formed. The σ factor is critical in RNAP binding to DNA as it is responsible for promoter recognition and binds to the -10 / -35 sites, thereby recruiting RNAP to the promoter. In bacterial cells σ are classified into two distinct families, with several species also encoding alternate σ factors that regulate specific sub-sets of genes in specific conditions. When the holoenzyme is bound to a promoter, the association of RNAP + σ factor + DNA is termed the closed complex. Another role of the σ factor is in facilitating the formation of the open complex (also referred to as the transcription bubble) by stabilizing unwound DNA.
In addition to σ factors, transcription factors (TF) are regulators important in controlling gene expression. TF are regulatory proteins that bind to specific sites, called transcription factor binding sites (TFBS), near genes to control transcription. Unlike σ factors, TF are able to promote or repress gene expression depending on the location of the TFBS in the promoter as represented in the figure below. Each TF regulates a network of genes in response to environmental signal or intracellular cues. This provides a mechanism by which bacterial cells are able to control a suite of genes all involved in the same cellular process or response. To illustrate the importance of TF in bacterial gene regulation, E. coli is predicted encode genes for ~270 TF. This accounts for 6% of the protein-coding genes in the genome! Though much of the current literature states that transcription factors only bind to the promoters of genes to regulate gene expression, recent research shows that these regulators also bind within the coding regions of gene sequences.
Protocols
Part 1: Examine targeted gene sequences for regulatory binding sites
Today you will examine the DNA sequences of the genes you targeted to identify features that may impact sgRNA_target binding. To do this you will first identify the -10 / -35 sites to define the promoter and second, identify any transcription factor binding sites (TFBS) within the promoter or gene sequence of the gene you targeted. The goal for this exercise is to identify if the CRISPRi system, guided by the sgRNA_target sequences, is interfering with the binding of regulatory proteins to the target site of the gene you selected. If so, this could provide information regarding the mechanism of gene regulation observed in your ethanol / acetate yield data. Furthermore, it could provide insight into how the ethanol or acetate yield can be improved.
- Use the KEGG Database to obtain the DNA sequences of the targeted genes in the E. coli K-12 MG1655 strain.
- Enter the name of gene you targeted in the Search genes box and click Go.
- Because some of sgRNA_target molecules were generated to target the promoter, enter 50 in the +upstream box to get the 50 basepair sequence immediately preceding the start codon.
- You can also use the GeneSnap file you created previously.
- First, examine the portion of the sequence that is the promoter and identify the -10 / -35 sites.
- You can do this visually using the image to the right as a guide or you can use BPROM, an online promoter prediction tool.


- In your laboratory notebook, record if your sgRNA_target sequence binds to the predicted -10 / -35 sites in the promoter, to another location in the promoter, or within the gene sequence. Create a schematic that shows the relative position of the sgRNA binding site in relation to the promoter / gene sequence.
- Second, examine the entire sequence and identify TFBS.
- Create a text file of your gRNA sequence + 25 base pairs both upstream and downstream of where your gRNA binds. You will use this sequence to identify any transcription factors with which your gRNA binding could interfere.
- Go to the Virtural Footprint interface and select the "Promotor analysis" option.
- In the section labeled 'Sequence', select the Paste option and insert the target region sequence you previously identified. To make this FASTA format compliant, include '>first' and hit Enter prior to the beginning of the sequence.
- In the section labeled 'Matrix' for the Position Weight Matrix, select E. coli transcription factors. You can select multiple transcription factors, but you must select each one manually. Although there are close to 100 E. coli transcription factors in the database, we have curated a smaller list for you to consider, linked here. Consider dividing the workload between lab partners.
- Click the 'Submit' button.
- The the new screen shows a table indicating the following information: 1. the basepair position of the start of the predicted TFBS, 2 on which strand the sequence is located (+ = nontemplate, - = template), 3. the TF predicted to bind at the identified binding site (more description on PWM is below), and the sequence of the predicted TFBS (5' → 3') 4. the score associated with the predicted TFBS (more description is below).
- In your laboratory notebook, record if your sgRNA_target sequencess binds to a predicted TFBS.
- Be specific as to which strand the sgRNA_target binds and which strand the TFBS is located.
- To better understand the predictions from the Virtual Footprint tool, let's look more closely at what the results mean for the TFBS identified using this analysis. For demonstration purposes, let's look at the results a random gene sequence:
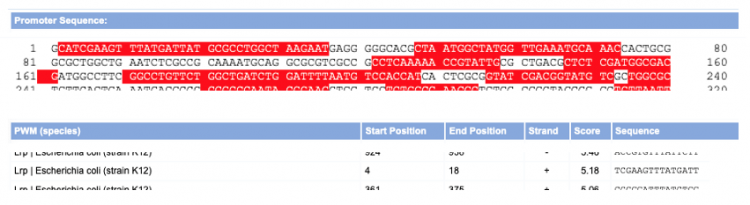
- The above screenshot shows a portion of the data for a random gene. The TFBS that we will investigate further is the site predicted for Lrp binding. Lrp is predicted to bind the template strand of the sequence entered from basepair 4 to 18.
- To begin, PWM refers to the position weight matrix. The PWM is used to represent the similarity to a DNA pattern and is constructed using the alignments of known binding sites.
- The PWM for the TFBS can be found by returning to the Virtual Footprint interface and selecting "Matrix list" at the top of the page.
- Select your transcription factor of interest and click on the link. A new window with more information will open.
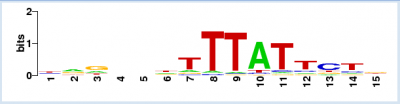
- The PWM for Lrp is shown to the right (this is located at the bottom of window that opens when you complete the above step). The size of each letter indicates the level of conservation at that base across the aligned sequences used to generate the PWM.
- The PWM for the TFBS can be found by returning to the Virtual Footprint interface and selecting "Matrix list" at the top of the page.
- The PWM is used to generate a score for each predicted TFBS in the sequence submitted for analysis.
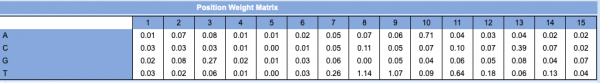
- A 'Position Weight Matrix' table for the Lrp PWM is shown to the right (this is located at top of the window with the PWM).
- This table contains values that indicate the level of conservation at each base across the aligned sequences used to generate the PWM.
- For the Lrp TFBS predicted in the random sequence use for this example, the first base is T. According to the PWM table, the numerical value for a T at position 1 of the TFBS = 0.01. If we did this for every base in the 15 TFBS sequence, the score would equal 5.18. This corresponds to the score provided in the results from the initial Virtual Footprint analysis (see image at Step #9). This is how the PWM score was calculated for the TF hits you identified. The 'core score' in parentheses is calculated by computing any possible six consequtive nucleotides for the region. The one with the highest value is labeled as the core score.
- In your laboratory notebook, complete the following for each TFBS identified in Step #8:
- Research the functional role of the TF that binds at that site. This can simply be a Google search that identifies the environmental conditions or intracellular cues under which it regulates gene expression.
- Include a screenshot of the PWM. Comparing the predicted TFBS sequence identified in your gene to the PWM for that TFBS, are you confident that this is a true binding site for the TF?




Next day: Organize figures and outline text for Research article