20.109(S22):M1D8

Contents
Introduction
Today is the final laboratory session for Module 1! You have completed all of the bench work for your research; however, there is still data analysis to complete for your experiments. In addition to plotting the data, you will complete statistical analysis to determine the significance of your results.
Statistics are mathematical tools used to analyze, interpret, and organize data. The specific tools that you will use are confidence intervals (CI) and the Student's t-test. To begin, review the following definitions:
- Mean (or average) is defined as:
$ \overline{\chi } = \frac{\sum_{i}^{n}\chi _{i}}{n} $, where $ \chi _{i} $ = individual value and n = number of samples
- With infinite data, the mean ($ \overline{\chi } $) approaches the true mean (μ).
- Standard deviation measures the variation in the data and is defined as:
$ s = \sqrt{\frac{\sum_{i}^{n }(\chi _{_{i}}-\overline{\chi })}{n - 1}} $, where n - 1 = degrees of freedom
- With infinite data, the standard deviation (s) approaches the true standard deviation (σ).
Because standard deviation is only justified when sufficient data have been collected to generate a normal curve, you will use confidence intervals to report the likelihood that your results predict the true mean. A confidence interval is a defined interval that is calculated to define the true mean to a specified level of confidence. Simply, it is possible to define a range in your data set that likely contains the true mean based on the calculated mean.
- Confidence interval (CI) is defined as:
CI = $ \overline{\chi } \pm \frac{ts}{\sqrt{n}} $, where t = value from t table (dependent on specified confidence level and n)
In your data, you should use the CI to generate error bars due the low n. Be sure to report which confidence level was used to calculate the intervals reported. So, what does this all mean in regard to the data you will report? As an example, if the calculated $ \overline{\chi } $ of a data set equals 80 au there is a 95% chance the μ is between 50 au and 110 au, where au = arbitrary units. And how does this relate to s? If you know the μ, the σ represents a 68% confidence interval.
When interpreting data, the error bars are representative of the noise in the data or how different the data points are for each of the replicates. Replicates come in two types: technical and biological. Technical replicates indicate that the same sample was tested multiple times and is measure of experimenter error (for example, pipetting errors between aliquots). Biological replicates indicate that different preparations of the same sample were tested and is a measure of the difference in a response to a variable (for example, response to a treatment between separate cultures of the same cell line). Though both types have value in data analysis, the interpretation of the error represented in each case is different. Because of this it is important to indicate if the replicates used in the data analysis are technical or biological. For your data, what type of replicates did you analyze for the γH2AX experiment? For the CometChip experiment?
Lastly, you will use Student's t-test to report if your data are statistically different between treatments.
- Student's t-test is defined as:
$ t = \frac{\left | \overline{\chi_{_{1}}} - \overline{\chi_{_{2}}} \right |}{s_{pooled}}\sqrt{\frac{n_{1} n_{2}}{n_{1}+n_{2}}} $, where $ s_{pooled} = \sqrt{\frac{s_{1}^{2} (n_{1} -1) + {s_{2}^{2} (n_{2} - 1)}{}}{n_{1} + n_{2} - 2}} $
The value you calculate with the Student's t-test equation is referred to as tcalculated. This tcalculated value is compared to the ttabulated value in the the t table, according to the appropriate n - 1 using the p-value for the two-tailed distribution (which assumes that you do not know how the data will shift). If the tcalculated value is greater than the ttabulated, then the data sets are significantly different at the specific p-value. So, what does this all mean in regard to the data you will report? As an example, if the tcalculated for a data set with n - 1 = 10 is 3 (given that the ttabulated is 2.228), then the data sets are different with a p-value ≤ 0.05. Which means that there is less that a 5% chance that the data sets are the same.
Protocols
Part 1: Image and analyze TDP43 localization experiment results

In fluorescence microscopy the specimen is illuminated with a wavelength of light specific to the excitation of the fluorescent tag used to target the feature of interest. The excitation wavelength is absorbed by the fluorescent tag, which causes it to emit light at a longer, less energetic wavelength. Typically, fluorescence microscopes used in biology are an epifluorescence type with a single light path (the objective) for excitation and emission detection, as depicted in the diagram above.
Fluorescence, or epifluorescence, microscopes are composed of a light source, an excitation filter, a dichroic mirror, and an emission filter. The filters and the dichroic mirror are specific to the spectral excitation and emission characteristics of the fluorescent tag. To visualize fluorescence, light at the excitation wavelength is focused on the sample. The light emission from the sample is focused by the objective to a detector.
To ensure you are familiar with the steps involved in imaging the TDP43-localization experiment, please watch the video tutorial linked here: [Imaging].
For timing reasons, the slides you prepared in the previous laboratory session were imaged prior to class today. The Instructors will provide a demonstration of the imaging process at the microscope so you understand how the data were captured for the analysis steps you will complete below. Please obtain the raw TDP43 images from the dropbox folder on the class data page. Three sets of images (i.e. image stacks) were taken per experimental condition, and each image stack contains images from two channels: DAPI (blue) and FITC (green). Remember that the secondary antibody used for the TDP43 staining was conjugated to an Alexa488 fluorophore, which emits green light. For each image stack, you will use ImageJ to 1) identify the location of the nuclei using the DAPI channel and 2) quantify the total TDP43 fluorescence in the FITC channel at locations specified by the DAPI channel.
Qualitatively assess signal localization and quantify total FITC intensity for all images
First, you will identify the total FITC signal intensity for each image.
- Open ImageJ.
- Go to Analyze -> Set Measurements.
- In the Set Measurements window, make sure the following boxes are checked: Area, Mean gray value, Min & max gray value, Shape descriptors, Integrated density, Display label.
- Go to Analyze -> Set Measurements.
- Open one image stack from the no treatment condition.
- The first image you see is the DAPI channel
- If you scroll to the right, the second image in the stack is the FITC (TDP-43) channel.
- Using the DAPI channel to orient yourself, examine the FITC signal localization for the image and use the questions below to note your qualitative assessment of the signal localization.
- While on the FITC channel image, Go to Analyze -> Measure
- A data box with assorted measurements will open. Copy the column headings and the data into an Excel spreadsheet to save for later use.
- Close the image and data box and repeat the process for the remaining images. This is your total measure of FITC intensity.
In your laboratory notebook, complete the following:
- What is your qualitative assessment of the CAD cells and TDP-43 localization?
- Are all nuclei approximately the same size and shape?
- Can you identify FITC signal in the nuclei (i.e. does the FITC channel have signal in roughly the same locations as the DAPI channel)? Is there FITC signal outside the nuclei? Where is most signal localized?
Identify intensity thresholds for DAPI channel

Next, you will identify intensity thresholds that will properly identify the cell nuclei in all the images. To be consistent and fair in analyzing fluorescence images, it is good practice to use the same intensity thresholds on all the images.
- Open ImageJ.
- Open one image stack from the no treatment condition.
- The first image you see is the DAPI channel
- If you scroll to the right, the second image in the stack is the FITC (TDP-43) channel.
- While the image is on the DAPI channel, go to Image -> Adjust -> Threshold.
- A threshold window should pop up
- Check the box for "Dark Background"
- Make sure the cell nuclei are highlighted in red.
- Adjust the threshold values to properly identify the majority of the cells' nuclei.
- Record the threshold values.
- Repeat this process for one image from each condition and cell line, and settle on threshold values for the DAPI channel that you will then use to analyze all the images. Write these values in your notebook.
- It is best to define the lower threshold value based on your images, and set the upper threshold value as 65535, which is the maximum possible intensity value for a 16-bit image.
- You can type in threshold values by clicking on the "Set" button in the Threshold window.
- Close all open images (File -> Close All).
Test TDP-43 nuclear quantification on one representative image
- In ImageJ, open one image to test the FITC quantification protocol.
- Split the image stack into two separate images.
- Go to Image -> Stacks -> Stack to Images.
- The DAPI image will have "-0001" as a suffix in its title.
- The FITC (TDP-43) image will have "-0002" as a suffix in its title.
- Duplicate the DAPI image and turn it into a mask to identify nuclei locations.
- Click on the DAPI image.
- Go to Image -> Duplicate, and click OK on the default title.
- Set the thresholds you chose on the duplicated DAPI image to identify nuclei.
- Go to Image -> Adjust -> Threshold.
- Check the box for "Dark Background".
- Click on the "Set" button and type in your threshold values (use 65535 for the upper threshold level).
- Go to Process -> Binary -> Convert to Mask.
- This makes the image black and white, where the white areas should correspond to nuclei locations.
- Use the newly created mask to identify locations on the FITC channel in which to quantify the TDP-43 signal in the nucleus.
- Go to Analyze -> Set Measurements.
- In the Set Measurements window, make sure the following boxes are checked: Area, Mean gray value, Min & max gray value, Shape descriptors, Integrated density, Display label.
- In the "Redirect to" field, scroll and select the FITC image (suffix -0002). Then press OK.
- This will direct ImageJ to the FITC image to analyze the metrics you selected in the areas identified by your mask. This will give you information about the TDP-43 signal in each nucleus.
- Go to Analyze -> Set Measurements.
- Run the analysis by selecting Analyze -> Analyze Particles.
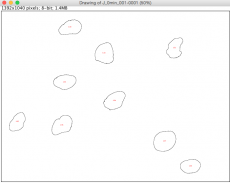 Example of areas identified by intensity thresholds for DAPI channel in ImageJ
Example of areas identified by intensity thresholds for DAPI channel in ImageJ
- In the "Size" field, type 200-Infinity. This will eliminate small, extraneous particles that do not correspond to nuclei.
- "Circularity" can remain at default values: 0-1.
- "Show" should say "Outlines".
- Click the following options: Display results, Exclude on edges, Summarize.
- Press OK to complete.
- A window will pop up showing outlines of each nucleus the software identified based on the thresholds you defined. Each identified area is labeled with a red number, corresponding to the left column of the data shown in the "Results" window.
- Take a look at the "Results" window to see the results of the analysis. It is good practice to validate the numerical results by comparing them to what you see in the images.
- The definition of the various measurements performed can be found on the ImageJ website (linked here).
- Does the nucleus with the largest "Area" correspond to the biggest nucleus you see in the drawing? The area here is in units of square pixels.
- The RawIntDens field is the total intensity (sum of the intensity of all the pixels) of the corresponding region. Does a region with a high total intensity value correspond to a cell with a high TDP43 signal? Click on the FITC image to double check.
- Close the "Results" window and do not save the data, as you will run the analysis on all the files together next.
- Close all open windows in ImageJ (File -> Close All).

Quantify nuclear TDP-43 signal in all images
- Create three folders on your desktop for each experimental condition: DMSO, 3uM SM, 30uM SM.
- Move the image files from both experimental dates into the appropriate folder according to the experimental condition in the file name.
- Ensure that all of your images from one experimental condition are in one folder together.
- Download AnalyzeNuclearStaining_Sp22 script (linked here).
- Right click on the link and download the file into a folder where you can find it.
- In ImageJ, go to Plugins--> Macros--> Run, and click on the AnalyzeNuclearStaining_Sp22 script that you downloaded.
- When the script prompts you to "Choose input folder," choose the folder containing all your .tif image stacks (folder named by one experimental condition), and click "Open."
- In the dialog box titled "Choose Intensity Threshold Values," type in the corresponding DAPI threshold values you have chosen, and click "OK."
- You will be prompted to name the resulting Excel file next.
- Please wait for the script to run through all your images for one condition. In the end all the image files will pop up, along with the "drawings" that show where it identified cells in your images.
- The script will output one Excel file into your image folder.
- Before closing any images, validate the results in the Excel file with the images in ImageJ.
- Choose a few representative images to verify.
- Check the "Drawing" images and DAPI images to see if the nuclei were called correctly by your threshold values.
- Repeat this analysis for each folder containing images from an experimental condition.
- To calculate total intensity of FITC signal per nucleus divide the RawIntDens (sum of the intensity of all the pixels in arbitrary units, a.u.) by area (pixels2) for each outline that represents a nucleus.
- Finally, calculate the average of the total FITC intensity(a.u.) per area (px2) for all nuclei in each experimental condition.
Compare cytoplasmic and nuclear TDP-43 signal across all treatment groups
- Average the nuclear FITC signal for all nuclei in a single image and record that value.
- Using the numbers you generated for the total and nuclear FITC signal for each image, calculate the percentage of FITC signal in the nuclei and non-nuclei regions of the cell for each image of each treatment.
Part 2: Use statistics to interpret results
Use the statistical analysis tools described in the Introduction to analyze the data for your aggregation and localization experiments. The figures / analyses in your Data summary should include measures of variability (i.e. confidence intervals) and significance (i.e. p-values).
For the aggregation data:
In the aggregation experiment that you completed, you included two replicates for each condition. For the figure that you will include in the Data summary, plot the averaged OD462 values then perform statistical analysis to determine the variability and significance of your data.
For the localization data:
In the localization experiment that you completed, you included replicates images from each coverslip for each condition. For the figure that you will include in the Data summary, plot the averaged FITC intensity values then perform statistical analysis to determine the variability and significance of your data.
In your laboratory notebook, complete the following:
- Include the plots for the aggregation and localization data with statistics indicated.
Next day: Complete in-silico cloning of pdCas9 expression plasmid