20.109(S21):M2D6

Introduction
Interactions between low molecular weight ligands and proteins have been shown to increase the thermostability of proteins. This means that proteins bound to ligand are able to maintain tertiary structure, or resist denaturation, at higher temperatures than unbound proteins. This concept is illustrated in the image below. Panel A shows that the native folded protein (blue solid line) denatures at a lower temperature than the native folded protein associated with drug (red dotted line).
The cellular thermal shift assay (CETSA) is a cell-based, or in-vivo, method used to identify low molecular weight ligands that bind and stabilize a protein of interest. In this assay, protein denaturation is measured by visualizing the presence or absence of protein after heat treatment. As shown in Panel B of the image below, samples from the native folded protein (top, blue lines) across different temperatures show that the protein denatures at a lower temperature than the native folded protein associated with drug (bottom, red lines). The presence of red bands at higher temperatures than blue bands indicates that the protein is stabilized when associated with the drug. More broadly, this result suggests that the drug does indeed associate with the protein of interest. Remember, the SMM screen identifies possible ligand:protein interactions. Therefore the CETSA method can be used to support the results of the SMM screen.
In that the cellular thermal shift assay uses whole cells to test protein stabilization in response to increasing temperature, this in-vivo test may provide information on the physiological relevance of protein:ligand binding. More specifically, the CETSA asks if a ligand is able to bind in a complex system (whole cell) whereas other in-vitro binding experiments test binding in an artificial system (reaction tube).

Protocols
Part 1: Select ligands that will be used for CETSA experiment
In total, five small molecules were assessed using CETSA. Review the information in the table below and select two small molecules for which you will analyze the CETSA data. When choosing molecules, consider the following:
- Review the groupings that you assigned based on the features in the small molecule hits that you identified from the SMM analysis.
- Do any of the small molecules that were examined via CETSA contain the features that you identified when grouping the hits that were identified in the SMM screen you completed?
- Use the SMILES information to determine which of the hits identified from the SMM screen were used for the CETSA experiments.
- Are there common features that can be identified among the five small molecules that were examined using CETSA?
- Because you will select two molecules, think about an interesting narrative for your Research article. What might be interesting about choosing two small molecules that contain the same feature? What might be interesting about choosing two small molecules that do not have similar features?
These online resources may be helpful to learning more about the hits that were identified in the SMM:
- Cloud version of ChemDraw here.
- Copy and paste the small molecule smiles into the work space to get a chemical structure
- Platform to transform the smiles information into a PubChem ID here.
- Copy and paste the smiles into the input ID search to determine the ID number.
- PubChem database of chemical information here.
- Includes small molecule molecular weight and other useful information.
In your laboratory notebook, complete the following:
- Record PubChem ID number and the formula for the two small molecules that you will analyze.
- From the prompts above, or any other reasoning that was used in your selection process, provide a brief rationale for why you chose to analyze those specific small molecules.
| Compound ID | SMM ID | Formula | Molecular weight | SMILES |
| 95877382 (2) | 01:KI0000454:E08 | C17H18N4O4S | 374.4 | S(=O)(=O)(c1ccc(cc1)CCNC(=O)c1cc(n[nH]1)c1oc(cc1)C)N |
| 69269200 (3) | 05:KI0001106:N03 | C17H23N3O2 | 301.4 | c1(N2C[C@H]([C@@](CC2)(O)CC)O)nc2c(cc1C#N)CCCC2 |
| 83079118 (4) | 13:KI0001562:P15 | C16H15FN4O | 298.3 | n1(c(n[nH]c1=O)Cc1ccncc1)[C@@H](c1ccc(cc1)F)C |
| 83023303 (5) | 02:KI0001354:N19 | C16H26N2O2 | 351.3 | C1(O)(CNCCC1)CNCCc1ccc(cc1)OCC |
| 26408703 (6) | 07:KI0000907:G08 | C18H27N7O2 | 373.5 | c12n(c(c(c(n1)C)CCC(=O)N1[C@H](C(=O)NC(C)C)C[C@@H](C1)N)C)ncn2 |
Part 2:Treat cells for CETSA experiment
Before you start your experiment, you need to calculate the dilutions that will be used for your treatment conditions. All of the ligands were prepared at two stock concentrations for this experiment, which will allow you to test each ligand at two treatment conditions. Use the following information (and the table below) to calculate the volume of each component should be added for each condition:
- Ligand stock concentrations = 1 mM and 10 mM
- You will use the 1 mM stock to prepare the 3 μM experimental treatment condition and the 10 mM stock to prepare the 30 μM treatment condition.
- To account for possible effects of DMSO (the ligands were dissolved in DMSO), the volume of DMSO added to the control should be equivalent to the volume of ligand added for the experimental treatments.
| Condition | DMSO | ligand (stock concentration = 1 mM) | ligand (stock concentration = 10 mM) |
| DMSO negative control | |||
| ligand treatment concentration = 3 μM | |||
| ligand treatment concentration = 30 μM |
- In your laboratory notebook, complete the following:
- Calculate the volumes of DMSO and / or ligand that will be added to each condition (you can include the above table).
- Obtain an aliquot of serum-free media and four 15 mL conical tubes.
- Add 10 mL of serum-free media to each conical tube and label accordingly:
- DMSO control
- 3 μM ligand
- 30 μM ligand
- Add the appropriate volume (calculated above) of each component to the correct conical tube. Be sure to double-check with your labels!
- Collect the four T75 flasks marked with your team color from the 37 °C incubator.
- Use the microscope to examine your cell cultures.
- Clearly label your flasks to reflect which you will use for the the experimental conditions (3 μM ligand and 30 μM ligand) and which you will use for the control condition (DMSO).
- Aspirate the media from the cells using a sterile Pasteur pipet.
- Wash the cells by adding 5 mL PBS using a 5 mL pipet. Slightly tip the flask back and forth to rinse the cells then aspirate the PBS with a Pasteur pipet.
- Add the correct serum-free media preparations to each of the flasks.
- Be sure to double-check that the serum-free media preparation added matches the label on the flask!
- Return your flasks to the 37 °C incubator for 1 hr.
- Following the incubation, you can take the cell cultures from the tissue culture room to your bench and complete the remaining steps in the main laboratory.
- Aspirate the serum-free media containing the ligand from the flasks.
- Wash the cells by adding 5 mL PBS using a 5 mL pipet. Slightly tip the flask back and forth to rinse the cells then aspirate the PBS with a Pasteur pipet.
- With a 2 mL pipet, add 1 mL of trypsin to each flask.
- Tip the flasks in each direction to distribute the trypsin evenly then incubate the cells at 37°C for 2 min.
- Retrieve your flasks from the incubator and firmly tap the bottom to dislodge the cells.
- Check your cells using the microscope to ensure they are dislodged. They should appear round and move freely.
- If your cells are not detached from the flasks, incubate at 37 °C for an additional minute.
- When your cells are dislodged, and add 3 mL of media to the cells then pipet the liquid up and down (“triturate”) to break up cells that are clumped together and suspend them in the liquid.
- Note: do not take up or release all the liquid, in order to avoid bubbles.
- Transfer the suspended cell cultures into separate, labeled 15 mL conical tubes.
- Pellet the cells for 3 min at 300 g in the centrifuge.
- Resuspend the cells in 3 mL PBS, then pellet for 3 min at 300 g in the centrifuge.
- Obtain an 1.5 mL aliquot of PBS containing protease inhibitor.
- Resuspend each cell pellet in 250 μL of PBS containing protease inhibitor.
- Transfer 100 μL of the DMSO-treated cell suspension to 2 labeled PCR tubes.
- One will be the unheated control and one be heated.
- Transfer 100 μL of each 'experimental' suspension to 1 labeled PCR tube.
- Both of the experimental samples will be heated to test for changes in protein stabilization in response to the ligand!
- The samples will be heated using the thermocyler. Take your tubes to the front laboratory bench and when all groups are ready, the samples will be heated for 3 minutes at 59 °C then cooled for 3 minutes at 25 °C.
- The teaching faculty will assist you as you complete the following steps.
- To snap freeze your cells, carefully drop your tubes in the liquid nitrogen for ~10 seconds.
- Carefully remove the tubes and place them in the thermocycler to thaw for 3 minutes at 25 °C.
- Complete the snap freeze procedure a total of three times; however after the third snap freeze, place the tubes in the -80 °C freezer instead of performing a final thaw. The samples will be stored at -80 °C until the next step.
Part 3: Prepare cellular proteins for imaging
To complete the CETSA experiment, the heat-treated cells need to be lysed and the lysate electrophoresed so the samples can be tested for the presence of native folded TDP43-RRM12 using a Western blot. Several steps need to be completed as part of this assay. First, steps similar to those completed previously in this module will be used. Recall that cellular proteins were separated using SDS-PAGE to examine the purity of the TDP43-RRM12 protein sample that was used for the SMM screen. Here we will again use SDS-PAGE to separate the proteins present in the heat-treated samples. Unlike the previous application of SDS-PAGE, we are now looking for the presence and abundance of just TDP43-RRM12. To do this we will an antibody specific for TDP43. Because we are probing for the presence and abundance of TDP43-RRM12 using an antibody, the proteins from the polyacrylamide gel must be transferred onto a nitrocellulose membrane that can be manipulated.
Lyse cells
- Retrieve your samples from the front laboratory bench and place them in the 25 °C heatblock.
- Your samples will thaw in the block.
- When thawed, transfer to an ice bucket.
- Vortex your samples, then centrifuge the lysate at 20,000 g for 20 min in the 4 °C cold room.
- Alert the teaching faculty when you are ready to centrifuge.
- This step removes the aggregated protein from your samples!
- Following centrifugation, leave your samples on ice.
- Be careful not to disturb the pellet!
- Transfer 75 μL of the lysate into a fresh 1.5 mL microcentrifuge tube.
- This is the soluble protein fraction from your samples!
In your laboratory notebook, complete the following:
- Why is it critical to complete the centrifugation in Step #2?
Electrophorese cellular proteins
- Add 15 μL of Laemmli sample buffer to each of your samples.
- Heat for 10 min at 70 °C using the heatblock on the front laboratory bench.
- If there is significant condensation at the lid of your tubes after heating, spin your samples before loading.
- You will load all samples and include a molecular weight standard on either side of your samples in the gel.
- 2 pre-stained ladders will be used to track the migration of your samples through the polyacrylamide gel
- Load 2 μL of the Licor ladder in Lane 1.
- Load 40 μL of each sample into the polyacrylamide gel.
- Load 5 μL of the BioRad ladder after the last sample.
- Your samples will be electrophoresed at 200 V for 30-40 min.
- Following electrophoresis, use the green Bio-rad tool to carefully pry apart the plates that encase your polyacrylamide gel.
- Using wet gloves, transfer your polyacrylamide gel to a dish and add enough dH2O to cover the gel.
- Wash the gel for 5 min at room temperature on the rotating table.
- Empty the water from the dish in the sink.
- Be careful that the gel does not fall into the sink!
- Repeat Steps #12-13 a total of 3 times.
In your laboratory notebook, complete the following:
- Which proteins are electrophoresed in the polyacrylamide gel? Is only TDP43-RRM12 present in the sample? Are other cellular proteins present in the sample?
Transfer proteins to membrane
To ensure the steps included below are clear, please watch the video tutorial linked here: [Protein Gel Transfer]. The steps are detailed below so you can follow along!
- Carefully open the iBlot transfer stack and separate the top stack from the bottom stack (see image to right for iBlot transfer stack setup).
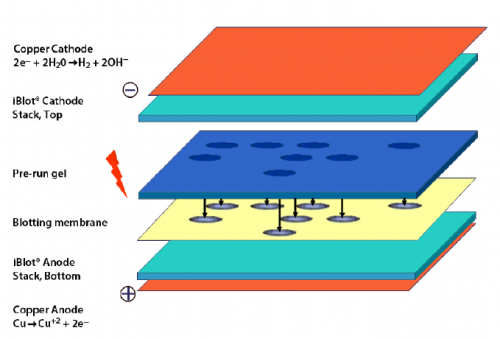

- Set it aside with the transfer gel layer facing upward.
- Keep the bottom stack in the plastic tray.
- Place the bottom stack with the plastic tray on the blotting surface of the iBlot device.
- Use the blotting roller to remove any air bubbles that are between the transfer stack and the membrane.
- Carefully place your polyacrylamide gel on the membrane.
- Two gels can fit onto one membrane, place each gel such that the ladder lane is along the bottom of the membrane and the top of the gel on the right.
- Use the blotting roller to remove any air bubbles that are between the membrane and the gel.
- Wet the filter paper in dH2O, then place on the gel.
- Use the blotting roller to remove any air bubbles that are between the gel and filter paper.
- Remove the plastic separator from the top stack.
- Place the top stack on the filter paper with the copper electrode facing upward.
- Place the absorbent pad on top of the transfer stack.
- Position the pad such that the electrical contacts (silver tabs) are aligned with the contacts on the blotting surface of the iBlot device.
- Close the iBlot device by gently pressing down on the two sides of the lid.
- Complete the transfer at 20 V for 7 min.
- Settings are saved as program P3 in the iBlot device.
- Carefully remove the transfer stack from the iBlot device and retrieve the membrane.
- Remember, the transfer step moves the proteins from the polyacrylamide gel onto the membrane!
- Place the membrane in a dish.
- Use one of your colored dot stickers to label the dish.
- Collect an aliquot of blocking buffer from the front laboratory bench and pour it over the membrane.
- Move the dish to the designated space in the 4 °C cooler.

In your laboratory notebook, complete the following:
- Why is it necessary to transfer the proteins electrophoresed in the polyacrylamide gel onto a nitrocellulose membrane?
Reagents list
- protease inhibitor cocktail (from Sigma)
- small molecule ligands (from Chembridge)
- 4-20% polyacrylamide gels in Tris-HCl (from Bio-Rad)
- TGS buffer: 5 mM Tris, 192 mM glycine, 0.1% (w/v) SDS (pH 8.3) (from Bio-Rad)
- ChameleonTM Duo Pre-stained Protein Ladder (from LI-COR)
- Molecular weights of ladder bands (linked here).
- 6x Reducing Laemmli Sample Buffer (from Boston BioProducts)
- iBlot 2 transfer stack (from ThermoFischer)
- Odyssey blocking buffer (from LI-COR)
Next day: Complete CETSA experiment and analyze data