20.109(F21):M1D3

Introduction
You will use commercially available antibodies to identify γH2AX foci in your experiment. The ability to bind specific proteins using antibodies, or immunoglobulins, is critical in immuno-fluorescence labeling analysis. Antibodies are typically 'raised' in mammalian hosts. Most commonly mice, rabbits, and goats are used, but antibodies can also be raised in sheep, chickens, rats, and even humans. The protein used to raise an antibody is called the antigen and the portion of the antigen that is recognized by an antibody is called the epitope. Some antibodies are monoclonal, or more appropriately “monospecific,” and recognize one epitope, while other antibodies, called polyclonal antibodies, are in fact antibody pools that recognize multiple epitopes. Antibodies can be raised not only to detect specific amino acid sequences, but also post-translational modifications and/or secondary structure. Therefore, antibodies can be used to distinguish between modified (for example, phosphorylated or glycoslyated proteins) and unmodified protein.
Monoclonal antibodies overcome many limitations of polyclonal pools in that they are specific to a particular epitope and can be produced in unlimited quantities. However, more time is required to establish these antibody-producing cells, called hybridomas, and it is a more expensive endeavor. In this process, normal antibody-producing B cells are fused with immortalized B cells, derived from myelomas, by chemical treatment with a limited efficiency. To select only heterogeneously fused cells, the cultures are maintained in medium in which myeloma cells alone cannot survive (often HAT medium). Normal B cells will naturally die over time with no intervention, so ultimately only the fused cells, called hybridomas, remain. A fused cell with two nuclei can be resolved into a stable cell line after mitosis.

To raise polyclonal antibodies, the antigen of interest is first purified and then injected into an animal. To elicit and enhance the animal’s immunogenic response, the antigen is often injected multiple times over several weeks in the presence of an immune-boosting compound called adjuvant. After some time, usually 4 to 8 weeks, samples of the animal’s blood are collected and the cellular fraction is removed by centrifugation. What is left, called the serum, can then be tested in the lab for the presence of specific antibodies. Even the very best antisera have no more than 10% of their antibodies directed against a particular antigen. The quality of any antiserum is judged by the purity (that it has few other antibodies), the specificity (that it recognizes the antigen and not other spurious proteins) and the concentration (sometimes called titer). Animals with strong responses to an antigen can be boosted with the antigen and then bled many times, so large volumes of antisera can be produced. However animals have limited life-spans and even the largest volumes of antiserum will eventually run out, requiring a new animal. The purity, specificity and titer of the new antiserum will likely differ from those of the first batch. High titer antisera against bacterial and viral proteins can be particularly precious since these antibodies are difficult to raise; most animals have seen these immunogens before and therefore don’t mount a major immune response when immunized. Antibodies against toxic proteins are also challenging to produce if they make the animals sick.

In your experiment, you will use a primary antibody to bind the γH2AX foci. Then a secondary antibody will be used that is specific to the conserved region of the primary antibody. The use of secondary antibodies allows researchers to tag the primary antibody. In our assay, the tag is a 488 nm fluorescent dye that will enable us to visualize double-strand breaks via microscopy. As a reminder, during the last laboratory session MCL-5 cells were treated with H2O2 +/- As for the γH2AX assay. Today we will discuss how to permeabilize the cells, which will enable the antibodies to enter the cells and bind γH2AX.
Protocols
Part 1: Participate in Communication Lab workshop
Our communication instructor, Dr. Prerna Bhargava, will join us today for a discussion on crafting data figures and legends.
Part 2: Perform antibody staining for γH2AX assay
Complete primary staining steps
Due to the timing of the As treatment, the Instructors completed the below steps using the coverslips that you prepared during the previous laboratory session. Using the these coverslips will complete the steps starting at Complete secondary staining steps.
To ensure the steps included below are clear, please watch the video tutorial linked here: [H2AX Staining]. The steps are detailed below so you can follow along!

- Obtain your 12-well plates from the front laboratory bench.
- Gather an aliquot of 1 X TBS from the front laboratory bench.
- Prepare 1.2 mL solution of 0.2% Triton X-100 (v/v) in 1X TBS in a micro centrifuge tube. 10% Triton stock is at the front laboratory bench.
- Prepare 2.5 mL solution of 1% BSA (v/v) in 1X TBS in 15ml conical tube. 10% BSA stock is at the front bench.
- One of the preparations will be the blocking solution used in Step #8 and the other preparation will be used in Step #9 for the primary antibody solution.
- Obtain a staining chamber from the front bench and add a damp paper towel to each side of the parafilm. Label parafilm with experimental details.
- Obtain a fine gauge (26 3/8) needle and a pair of tweezers from the front laboratory bench.
- Carefully press the tip of the needle against the benchtop to bend it into a right angle such that the beveled side of the needle is the interior angle.
- Use the 'hook' created with the needle to lift the coverslip from the bottom of the well, then use the tweezers to 'catch' the coverslip.
- Practice plates with coverslips will be available at the front laboratory bench.
- When you are confident with your ability to retrieve the coverslips from the wells, move one coverslip from each condition from your 12-well plates to the staining chamber. Cell-side UP!
- The cell-side of the coverslip is the side that was facing up in the well of the 12-well plate.
- Quickly permeabilize the cells by adding 150 μL of the 0.2% Triton X-100/TBS solution to each coverslip and incubate for 10 min at room temperature.
- Aspirate the 0.2% Triton X-100/TBS solution and add 150 μL of BSA blocking solution to each coverslip, then incubate for 60 min at room temperature.
- With 15 min remaining of the blocking solution incubation, prepare the primary antibody.
- Dilute the mouse anti-γH2AX antibody 1:1000 in the 1.2 mL aliquot of BSA blocking solution.
- Aspirate the block solution and add 150 μL of the diluted primary antibody solution to each coverslip before moving the next. Do not let the coverslips dry!
- Carefully move your staining chamber to the 4 °C cooler.
- Incubate samples at 4 °C in the primary antibody solution for ~48 h.
Complete secondary staining steps
- Retrieve the staining chamber with your coverslips from the 4 °C cooler.
- Wash each coverslip by pipetting 1 mL of TBS-Triton to the top of the coverslip, then use pipet to remove liquid.
- Complete a total of 3 washes. At the final wash leave the liquid on the coverslip.
- Dilute the secondary antibody, Alexa Fluor 488 goat anti-mouse, 1:200 in a 1.2 mL aliquot of blocking solution.
- Aspirate the wash liquid from one coverslip and immediately add 150 μL of the diluted secondary antibody to the coverslip.
- Complete this step for each coverslip individually as it is important that the coverslips do not dry!
- Carefully move your staining chamber to the 4 °C cooler.
- Incubate samples at 4 °C in the secondary antibody solution for ~1 h.
- Make sure to have TBS solution available before you start. Aspirate the secondary antibody solution off the coverslip and immediately add 150 μL of TBS. Do not let the coverslips dry out during this process.
- To complete the post secondary wash, add 150 μL of TBS per coverslip, let incubate at room temperature for 3 min covered, then aspirate.
- To add DAPI, dilute the DAPI stain 1:1000 in TBS and add 150 μL DAPI per coverslip. Let incubate at room temperature for 10 min covered, then aspirate.
- Add TBS as in step 2 for the final wash and leave for 3 min. Do no aspirate.
- Obtain glass slides from the front laboratory bench and label your slides with all of your experimental information and group name, add one drop of mounting media to the slide.
- Aspirate the final TBS wash and using tweezers place the coverslip cell-side down on the mounting media "spot" on the microscope slide. Try your best to avoid bubbles by slowly placing the coverslip over the mounting media.
- The cell-side of the coverslip is the side that was facing up in the staining chamber.
- Complete Steps #5-6 for coverslips from all of the coverslips you stained.
- Add one small drop of nail polish to each side of your coverslip to seal it to the glass slide.
In your laboratory notebook, complete the following:
- Why is it important to wash the secondary antibody from the coverslip before imaging?
- What stain is used following secondary antibody? What cellular component is stained in this step? And why is this useful?
Part 3: Image γH2AX experiment

In fluorescence microscopy the specimen is illuminated with a wavelength of light specific to the excitation of the fluorescent tag used to target the feature of interest. The excitation wavelength is absorbed by the fluorescent tag, which causes it to emit light at a longer, less energetic wavelength. Typically, fluorescence microscopes used in biology are an epifluorescence type with a single light path (the objective) for excitation and emission detection, as depicted in the diagram above.
Fluorescence, or epifluorescence, microscopes are composed of a light source, an excitation filter, a dichroic mirror, and an emission filter. The filters and the dichroic mirror are specific to the spectral excitation and emission characteristics of the fluorescent tag. To visualize fluorescence, light at the excitation wavelength is focused on the sample. The light emission from the sample is focused by the objective to a detector.
To ensure you are familiar with the steps involved in imaging the γH2AX experiment, please watch the video tutorial linked here: [H2AX Imaging].
Part 4: Analyze γH2AX images by measuring fluorescence intensity
Please obtain the raw γH2AX images from the Class Dropbox folder. Find your group subfolder which contains images taken from your coverslips stained by the teaching faculty. Six sets of images (i.e. image stacks) were taken per experimental condition, and each image stack contains images from two channels: DAPI (blue) and FITC (green). Remember that the secondary antibody used for the γH2AX staining was conjugated to an Alexa488 fluorophore, which emits green light. For each image stack, you will use ImageJ to 1) identify the location of the nuclei using the DAPI channel and 2) quantify the total γH2AX fluorescence in the FITC channel at locations specified by the DAPI channel.
Identify intensity thresholds for DAPI channel

First, you will identify intensity thresholds that will properly identify the cell nuclei in all the images. To be consistent and fair in analyzing fluorescence images, it is good practice to use the same intensity thresholds on all the images.
- Open ImageJ.
- Open one image stack from the no treatment condition.
- The first image you see is the DAPI channel
- If you scroll to the right, the second image in the stack is the FITC (γH2AX) channel.
- While the image is on the DAPI channel, go to Image -> Adjust -> Threshold.
- A threshold window should pop up
- Check the box for "Dark Background"
- Make sure the cell nuclei are highlighted in red.
- Adjust the threshold values to properly identify the majority of the cells' nuclei.
- Record the threshold values.
- Repeat this process for one image from each condition and cell line, and settle on threshold values for the DAPI channel that you will then use to analyze all the images. Write these values in your notebook.
- It is best to define the lower threshold value based on your images, and set the upper threshold value as 256, which is the maximum possible intensity value for a 8-bit image.
- You can type in threshold values by clicking on the "Set" button in the Threshold window.
- Close all open images (File -> Close All).
Test γH2AX quantification on one representative image
- In ImageJ, open one image to test the FITC quantification protocol.
- Split the image stack into two separate images.
- Go to Image -> Stacks -> Stack to Images.
- The DAPI image will have "-0001" as a suffix in its title.
- The FITC (gamma-H2AX) image will have "-0002" as a suffix in its title.
- Duplicate the DAPI image and turn it into a mask to identify nuclei locations.
- Click on the DAPI image.
- Go to Image -> Duplicate, and click OK on the default title.
- Set the thresholds you chose on the duplicated DAPI image to identify nuclei.
- Go to Image -> Adjust -> Threshold.
- Check the box for "Dark Background".
- Click on the "Set" button and type in your threshold values (use 256 for the upper threshold level).
- Go to Process -> Binary -> Convert to Mask.
- This makes the image black and white, where the white areas should correspond to nuclei locations.
- Use the newly created mask to identify locations on the Texas Red channel in which to quantify the gamma-H2AX signal.
- Go to Analyze -> Set Measurements.
- In the Set Measurements window, make sure the following boxes are checked: Area, Mean gray value, Min & max gray value, Shape descriptors, Integrated density, Display label.
- In the "Redirect to" field, scroll and select the FITC image (suffix -0002). Then press OK.
- This will direct ImageJ to the FITC image to analyze the metrics you selected in the areas identified by your mask. This will give you information about the gamma-H2AX signal in each nucleus.
- Go to Analyze -> Set Measurements.
- Run the analysis by selecting Analyze -> Analyze Particles.
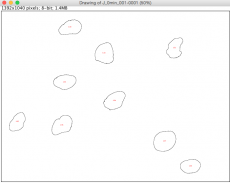 Example of areas identified by intensity thresholds for DAPI channel in ImageJ
Example of areas identified by intensity thresholds for DAPI channel in ImageJ
- In the "Size" field, type 200-Infinity. This will eliminate small, extraneous particles that do not correspond to nuclei.
- "Circularity" can remain at default values: 0-1.
- "Show" should say "Outlines".
- Click the following options: Display results, Exclude on edges, Summarize.
- Press OK to complete.
- A window will pop up showing outlines of each nucleus the software identified based on the thresholds you defined. Each identified area is labeled with a red number, corresponding to the left column of the data shown in the "Results" window.
- Take a look at the "Results" window to see the results of the analysis. It is good practice to validate the numerical results by comparing them to what you see in the images.
- The definition of the various measurements performed can be found on the ImageJ website (linked here).
- Does the nucleus with the largest "Area" correspond to the biggest nucleus you see in the drawing? The area here is in units of square pixels.
- The RawIntDens field is the total intensity (sum of the intensity of all the pixels) of the corresponding region. Does a region with a high total intensity value correspond to a cell with a high gamma-H2AX signal? Click on the FITC image to double check.
- Close the "Results" window and do not save the data, as you will run the analysis on all the files together next.
- Close all open windows in ImageJ (File -> Close All).

Quantify γH2AX signal in all images
- Create four folders on your desktop for each experimental condition: No treatment, 100uM H2O2, 2uM Arsenic and 2uM Arsenic+100uM H2O2.
- Move the image files from both experimental dates into the appropriate folder according to the experimental condition in the file name.
- Ensure that all of your images from one experimental condition are in one folder together.
- Download AnalyzeH2AX_FITCintensityBatch_Fa20script (linked here).
- Right click on the link and download the file into a folder where you can find it.
- In ImageJ, go to Plugins--> Macros--> Run, and click on the AnalyzeH2AX_FITCintensityBatch_Fa18 script that you downloaded.
- When the script prompts you to "Choose input folder," choose the folder containing all your .tif image stacks (folder named by one experimental condition), and click "Open."
- In the dialog box titled "Choose Intensity Threshold Values," type in the corresponding DAPI threshold values you have chosen, and click "OK."
- You will be prompted to name the resulting Excel file next.
- Please wait for the script to run through all your images for one condition. In the end all the image files will pop up, along with the "drawings" that show where it identified cells in your images.
- The script will output one Excel file into your image folder.
- Before closing any images, validate the results in the Excel file with the images in ImageJ.
- Choose a few representative images to verify.
- Check the "Drawing" images and DAPI images to see if the nuclei were called correctly by your threshold values.
- Repeat this analysis for each folder containing images from an experimental condition.
- To calculate total intensity of FITC signal per nucleus divide the RawIntDens (sum of the intensity of all the pixels in arbitrary units, a.u.) by area (pixels2) for each outline that represents a nucleus.
- Finally, calculate the average of the total FITC intensity(a.u.) per area (px2) for all nuclei in each experimental condition.
Part 5: Analyze γH2AX images by counting foci
In addition to the above analysis, you will also use ImageJ to enumerate the γH2AX foci present in the nuclei of the treated cells. To do this use the protocol written by researchers at Duke University. Follow the direction outlined here to analyze your images.
In your laboratory notebook, complete the following:
- How does the data obtained in the two analysis approaches compare? Are the results the same? Different?
- Which analysis approach best represents the raw data images? Why?
Reagents list
- permeabilization buffer: 0.2% Triton in Tris buffer saline (TBS) (from Invitrogen)
- blocking buffer: 1% bovine serum albumin (BSA) in TBS (BSA from Sigma)
- 1:1000 primary antibody to γH2AX, mouse (from Millipore)
- 1:200 Alexa Fluor 488 goat anti-mouse IgG (from ThermoFisher)
- 1:1000 DAPI (from ThermoFisher)
- Fluoromount G (from Southern Biotech)
Next day: Treat cells and perform high-throughput genome damage assay