20.109(F16):Design gRNA for CRISPRi (Day2)

Contents
Introduction
Brief CRISPR(i)...
Metabolic engineering...
Protocols
During your last laboratory session you completed an experiment to confirm the pdCas9 plasmid. Today you will examine the results of your digests and perform the next stage of preparatory work by selecting a target for your system engineering project. Upon determining which gene you will target, you will design a gRNA specific to your target.
Part 1: Agarose gel electrophoresis of confirmation digests
Electrophoresis is a technique that separates large molecules by size using an applied electrical field and a sieving matrix. DNA, RNA and proteins are the molecules most often studied with this technique; agarose and acrylamide gels are the two most common sieves. The molecules to be separated enter the matrix through a well at one end and are pulled through the matrix when a current is applied across it. The larger molecules get entwined in the matrix and are stalled; the smaller molecules wind through the matrix more easily and travel further from the well. The distance a DNA fragment travels is inversely proportional to the log of its length. Over time fragments of similar length accumulate into “bands” in the gel. Higher concentrations of agarose can be used to resolve smaller DNA fragments.

DNA and RNA are negatively charged molecules due to their phosphate backbone, and they naturally travel toward the positive charge at the far end of the gel. Today you will separate DNA fragments using an agarose matrix. Agarose is a polymer that comes from seaweed and if you’ve ever made Jell-O™, then you already have all the skills needed for pouring an agarose gel! To prepare these gels, agarose and 1X TAE buffer are microwaved until the agarose is melted and fully dissolved. The molten agar is then poured into a horizontal casting tray, and a comb is added. Once the agar has solidified, the comb is removed, leaving wells into which the DNA sample can be loaded.
You will use a 1% agarose gel (prepared by the teaching faculty) to separate the DNA fragments in your four digested samples as well as a reference lane of molecular weight markers (also called a DNA ladder).
- Add 5 μL of 6x loading dye to the digests.
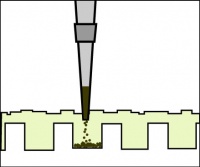 Illustration of proper gel loading technique.
Illustration of proper gel loading technique.
- Loading dye contains bromophenol blue as a tracking dye to follow the progress of the electrophoresis (so you don’t run the smallest fragments off the end of your gel!) as well as glycerol to help the samples sink into the well.
- Flick the eppendorf tubes to mix the contents, then quick spin them in the microfuge to bring the contents of the tubes to the bottom.
- Load 25 μL of each digest into the gel, as well as 20 μL of 1kb DNA ladder.
- Be sure to record the order in which you load your samples!
- To load your samples, draw the volume listed above into the tip of your P200 or P20. Lower the tip below the surface of the buffer and directly over the well. You risk puncturing the bottom of the well if you lower the tip too far into the well itself (puncturing well = bad!). Expel your sample into the well. Do not release the pipet plunger until after you have removed the tip from the gel box (or you'll draw your sample back into the tip!).
- Once all the samples have been loaded, attach the gel box to the power supply and run the gel at 125 V for no more than 45 minutes.

Part 2: Select E. coli fermentation pathway protein to target
Your goal in this module is to increase the production of either ethanol or lactate, two valuable products of the E. coli fermentation pathway. You will complete this task using the CRISPRi system, which targets genes such that transcription of the target gene is decreased, thus resulting in less of the protein encoded by the targeted gene.

Use the fermentation pathway from E. coli (included below) and the example above to answer the following questions with your partner.
- Which gene(s) might you target to increase the availability of substrate for ethanol production? For acetate production?
- Which gene(s) might you target to decrease the amount of substrate used to generate products in steps downstream of ethanol production? For acetate production?

With your laboratory partner, review Metabolic engineering of Escherichia coli for production of mixed-acid fermentation end products by Forster & Gescher. Using the information within this article and the genes that you identified above, select one gene that you will target in an attempt to increase the production of either ethanol or lactate. Be sure to include notes on your decision and thought process in your laboratory notebook.
Before you continue, note the fermentation product you aim to increase and the gene you will target in the Discussion tab.
Part 3: Design gRNA for CRISPRi system
Now that you know which gene you will target with the CRIPSRi system, you need to design a guide RNA (gRNA) using the DNA sequence. As a reminder, the gRNA recruits dCas9 to a specific location in the genome where it will bind and block transcription. The CRISPRi system can block transcription at either the initiation or elongation steps of transcription (see below image). If the dCas9 protein is bound to the promoter sequence at either a transcription factor binding site (TFBS) or the -10/-35 boxes it will block transcription factors and/or RNA polymerase from binding to the promoter of the gene, thereby impeding transcription initiation. If the dCas9 protein is bound within the gene it impede RNA polymerase elongation of the transcript. Consider both options with your partner and decide on a strategy before working through the remaining protocol. And, as always, record your thoughts in your notebook.

When you decide on which region of your target gene you will use to design your gRNA follow the steps below to choose a gRNA sequence.
- Use the KEGG Database to obtain the DNA sequence of your target gene in the E. coli K-12 MG1655 strain.
- Enter the name of your target gene in the Search genes box and click Go.
- Double click on the linked gene name.
- Using the information provided by the KEGG database, answer the following questions:
- What is the full name of your gene (or Definition)?
- In what pathways is your gene involved?
- Determine the position of your gene within the genome by clicking the Genome map button within the Position row.
- Is your target gene at the start of an operon or in the middle?
- Does the position of your gene within the operon affect the gRNA strategy you discussed with your partner? If you are unsure, consult with the teaching faculty.
- The amino acid (AA) sequence and nucleotide sequence (NT) of your gene is provided at the bottom of the page.
- Generate a new sequence file in Benchling that contains the NT sequence of your gene.
- If you will design a gRNA that binds the promoter, enter 50 in the +upstream box to get the 50 basepair sequence immediately preceding the start codon.
- Use the DNA sequence to select a 20-25 basepair region that will serve as the gRNA binding site.
- If you target the template DNA strand, the gRNA sequence will be the same as the transcribed sequence.
- If you target the nontemplate strand, the gRNA sequence will be the reverse-complement of the transcribed sequence.
- To avoid off-target effects, check the specificity of your gRNA sequence using the Basic Local Alignment Search Tool (BLAST) available from NCBI.
- Click on the Nucleotide BLAST icon.
- In the Enter Query Sequence section, paste your gRNA sequence into the Enter accession number(s), gi(s), or FASTA sequence(s) box.
- In the Choose Search Set section, type 'MG1655' in the Organism box and select an appropriate option from the drop down list.
- Click the BLAST button at the bottom of the screen.
- Ideally, your gRNA sequence will only have 100% identity to the target sequence.
- If your gRNA sequence binds to multiple locations (with a 14+ nucleotide region within the gRNA having 100% identity to an off-target) within the genome, consider redesigning your gRNA.
- When you are satisfied with your gRNA sequence selection, post it to the Discussion tab and complete the requested information in the table.
- Be sure you write your gRNA sequence 5' → 3'
An additional sequence 'tag' will be added to your gRNA. The teaching faculty will order your gRNA as a primer from Integrated DNA Technologies (IDT).
Reagents
Next day: Generate gRNA plasmid