20.109(S09):Induce protein and evaluate DNA (Day5)
Introduction
revise to add diagnostic digests as alternative/complement to sequencing information

.png)
Last time you transformed your mutant DNA into BL21(DE3) cells. The colonies that arose were moved to liquid cultures, and today you will add IPTG to these cultures to induce protein expression by the bacteria. Next time you will purify the resultant protein. I won’t shy away from telling you that there are many things that can go wrong at this stage! However, each one is certainly a learning experience.
As evidenced by Nagai’s work, wild-type inverse pericam is not toxic to BL21(DE3) cells. Although it is unlikely for your small mutation to dramatically change this fact, in general a novel protein may turn out to be toxic. If this is the case, only very small amounts of protein are produced before the bacteria die. Keep in mind that overexpressing a single protein may come at the expense of producing proteins needed for survival, and will most likely cause cell death eventually; however, toxic proteins hasten this demise. Aberrant toxicity can sometimes be alleviated by reducing the culture temperature (e.g., to 30 °C).
Based on its fluorescence activity, wild-type inverse pericam allows proper folding of (cp)EYFP, and based on its response to calcium, it also allows calmodulin to fold. One problem you may encounter is that your mutant proteins will no longer fold correctly. Since you made mutations in the calcium sensor part of IPC, rather than the fluorescent part, it is unlikely that your protein will destroy EYFP fluorescence. However, a common problem with misfolded proteins is the formation of insoluble aggregates, due for instance to improperly exposed hydrophobic surfaces. Proteins can be purified from these aggregates – called inclusion bodies – but the process is more labour-intensive than for soluble proteins. (The proteins must be extracted under more harsh conditions than you will use next time, then purified under denaturing conditions, before finally attempting to renature the proteins.) Inclusion bodies sometimes form simply due to very high expression of the protein of interest, causing it to pass its solubility limit. This outcome can be prevented by lowering the culture temperature or time, the amount of IPTG, or the growth phase of the bacteria.
One final point to keep in mind is that not all proteins can be produced in bacteria. Eukaryotic proteins that require post-translational modifications (such as glycosylation) for activity require eukaryotic hosts (such as yeast, or the ubiquitous CHO – Chinese hamster ovary – cells). Sometimes eukaryote-derived proteins will be truncated or otherwise mistranslated by E. coli due to differential codon bias; errors in translation can be prevented by providing additional tRNAs to the culture or directly to the bacteria via plasmids. Despite all this complexity, prokaryotic hosts have been plenty good enough to produce proteins for certain therapies, notably the cytokine G-CSF for patients needing to replenish their white blood cells (e.g., after chemotherapy), sold as Neupogen by Amgen.

After you induce your cells with IPTG, you will let the resultant protein factories do their work for 2-3 hours. During this time, you will look at the sequencing results for your mutant plasmids, to determine if they have the right mutation (and only that mutation). The invention of automated sequencing machines has made sequence determination a fast and inexpensive endeavor. The method for sequencing DNA is not new but automation of the process is recent, developed in conjunction with the massive genome sequencing efforts of the 1990s. At the heart of sequencing reactions is chemistry worked out by Fred Sanger in the 1970’s which uses dideoxynucleotides (see figure at right).
These chain-terminating bases can be added to a growing chain of DNA but cannot be further extended. Performing four reactions, each with a different chain-terminating base, generates fragments of different lengths ending at G, A, T, or C. The fragments, once separated by size, reflect the DNA’s sequence. In the “old days” (all of 10 years ago!) radioactive material was incorporated into the elongating DNA fragments so they could be visualized on X-ray film (image on left). More recently fluorescent dyes, one color linked to each dideoxy-base, have been used instead. The four colored fragments can be passed through capillaries to a computer that can read the output and trace the color intensities detected (image on right). Your sample was sequenced in this way on an ABI 3730 DNA Analyzer.
Sequencing gel
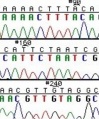
Sequence trace data


Analysis of sequence data is no small task. “Sequence gazing” can swallow hours of time with little or no results. There are also many web-based programs to decipher patterns. The nucleotide or its translated protein can be examined in this way. Thanks to the genome sequence information that is now available, a new verb, “to BLAST,” has been coined to describe the comparison of your own sequence to sequences from other organisms. BLAST is an acronym for Basic Local Alignment Search Tool, and can be accessed through the National Center for Biotechnology Information (NCBI) home page.