20.109(S21):M1D3

Contents
Introduction:Harvest clone plasmids from yeast library
Last time you sorted a population of yeast from a library using FACS based on their ability to bind to lysozyme, our ligand of interest. The cells were selected for having a green fluorescent signal correlated with surface expression of a single-chain antibody fragment (scFv) and a far-red fluorescent signal correlated with binding to lysozyme. The goal of today's experiment is to harvest the DNA plasmid expressing the unique antibody fragment from the mixed population of yeast. You will then transform that mixed population of "improved binder" plasmids from yeast into E. coli bacteria to amplify the plasmid.
After isolation of the plasmid, you will quantify your DNA by spectrophotometry. Finally you will transform the yeast plasmid DNA into E. coli. Yeast are not optimized to create many copies of plasmid DNA and we need a much higher concentration of DNA to analyze the sequence of the plasmid. Luckily for us engineered E. coli are relatively inexpensive and will quickly amplify plasmids. Most E. coli do not usually exist in a state in which they will easily take up plasmid DNA, but the bacteria can be prepared such that they are permeable to the plasmid DNA. The process of taking up foreign DNA into a cell is called transformation, and when cells are prepared to take up DNA they are called competent. The transformation procedure is efficient enough for most lab purposes, with efficiencies as high as 109 transformed cells per microgram of DNA, but it is important to realize that even with high efficiency cells only 1 DNA molecule in about 10,000 is successfully transformed. In our transformation protocol we will adjust the amount of plasmid DNA such that the E. coli will each take up between 0-1 plasmid per competent cell. The cells that do not take up plasmid DNA will not survive in the selective culture media (or on selective media plates). Selective media typically contains an antibiotic which kills microbes that do not produce the necessary enzyme to neutralize the antibiotic. The enzyme for a given antibiotic is encoded in the plasmid DNA. We can then harvest individual colonies of E. coli to find unique sequences of improved binders.

Protocols:
Part 1: Participate in Comm Lab workshop
Our communication instructors, Dr. Prerna Bhargava and Dr. Sean Clarke, will join us today for a discussion on crafting Figures and Captions.
Part 2: Harvest plasmid DNA from yeast

Motivation: To determine the sequences of the new scFvs with improved binding to lysozyme we must first harvest the plasmids from the yeast cells.
The yeast that were sorted via FACS in our last lab session were incubated at 30°C for an additional three days so the yeast could recover from the sorting, grow and divide. This additional growth time will give us a sufficient number of cells to harvest plasmid DNA.
To harvest plasmid DNA from yeast we need to do a bit more work to break down the cell wall. Yesterday the 2x107 yeast cells were pelleted and digested with zymolase overnight at 37°C. Zymolase is a mixture of enzymes purified from the bacteria Arthrobacter luteus that degrades the cell wall of yeast and other fungi. The yeast plasmid purification will be carried out using reagents from a commercial kit called Zymoprep MiniPrep II.
- Retrieve your overnight yeast digests from the 37°C incubator.
- Add 200uL of Solution 2 and vortex briefly.
- This is the alkaline buffer.
- Add 400uL of Solution 3, vortex briefly and centrifuge at 14,000g for 10min.
- This is the buffer that neutralizes the pH.
- Transfer the supernatant to a new 1.5 mL tube and centrifuge at 14,000g for an additional 10min.
- Transfer the supernatant to a blue DNA binding, silica column and centrifuge at 10,000g for 30sec. Discard the flow through into a tube labeled 'zymo waste'.
- Add 550uL DNA wash buffer to the column, centrifuge at 10,000g for 2min, and discard the flow through to 'zymo waste'.
- Transfer the column to a new 1.5 mL tube and carefully add 10uL of water to the center of the column to elute the DNA.
- Let the column sit for 2min at room temp.
- Centrifuge at 10,000g for 1min to elute the plasmid DNA from the column and collect the flow-through.
- Discard the column and keep the 1.5 mL tube with the plasmid DNA on ice.
Part 3: Using a spectrophotometer to measure DNA concentration
Motivation: The following bacterial transformations require a precise amount of DNA. We will use DNA spectroscopy to determine the concentration of DNA we purified from the yeast population.
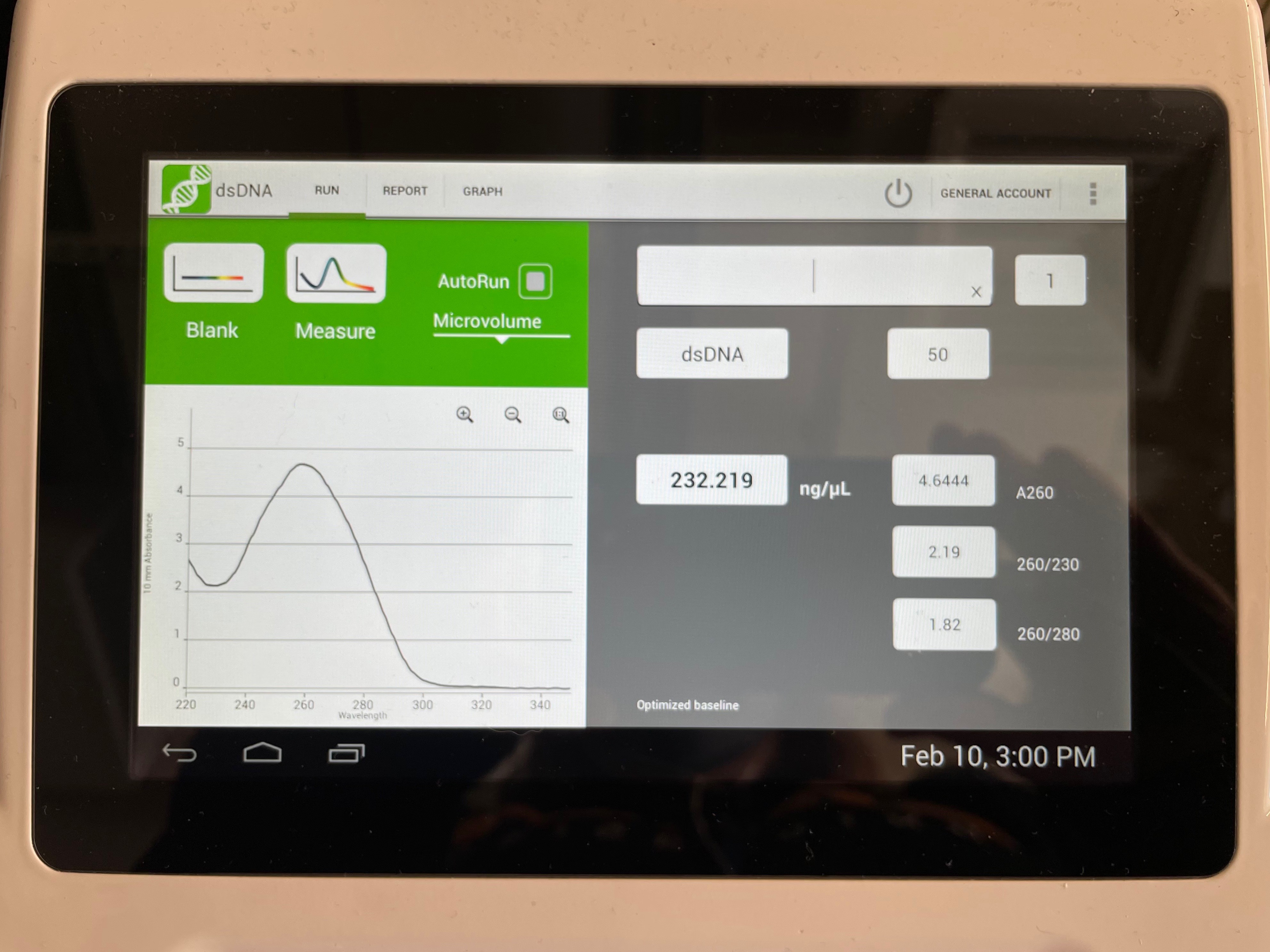
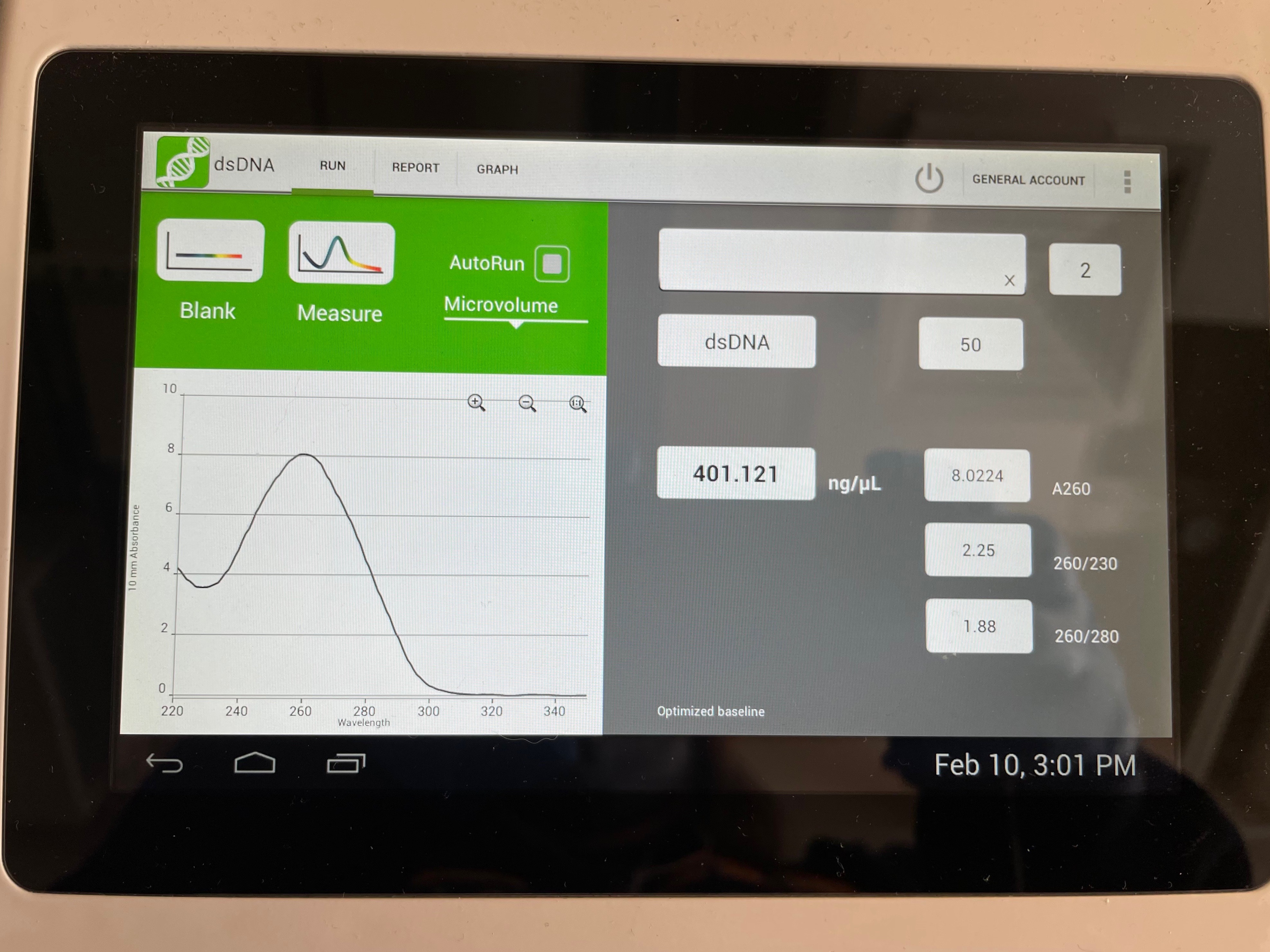
Nucleic acids (both RNA and DNA) have an absorbance peak at 260 nm. Beer's law may be used to quantify the amount of DNA from this peak: Abs = ε l c, where Abs is the measured absorbance, l is the path length (1 cm for most specs), c is concentration, and ε is the extinction coefficient. For DNA, ε is 0.02 (μg/mL cm)-1, so 1 absorbance unit corresponds to 50 μg/mL of DNA.
We also measure absorbance at other wavelengths to determine DNA purity. Proteins have absorbance peaks at 280 nm (primarily due to the aromatic peptides tryptophan and tyrosine). An Abs260:Abs280 ratio of ~1.8:1 is desired for pure DNA. Absorbance at 230 or less is usually the result of other impurities like phenol or guanidine buffers. An Abs260:Abs230 ratio of 2.0-2.2 is expected.
The advantage of using a nano drop to measure DNA is the small volume of sample that is lost during measurement. We can get very precise measurements from very small volumes of DNA. This is very important in our experiment because we want to retain as much of the purified library plasmids as possible for analysis.



- You will now take your samples to the Nanodrop to measure the DNA's absorbance to quantify the concentration.
- Select dsDNA.
- Add 2uL of water or the buffer you eluted DNA into for the blank.
- Close the arm over the spectrometer and make sure the liquid solution is bridging between the pedestal and arm.
- Press Blank.
- Open the arm and wipe the water off with a Kimwipe.
- Add 2uL of DNA to the pedestal.
- Select measure dsDNA button and record DNA concentration and purity.
- Open the spectrometer arm and wipe the pedestal with a Kimwipe.
- Wet a second Kimwipe with dH2O and wipe the pedestal again. Leave the arm up to air-dry.
In your laboratory notebook, complete the following:
- Open the following measurements: Spec1 and Spec2
- Which measurement has the highest concentration of DNA?
- Are both pure DNA samples? Explain.
- What volume would you use in Part 3 step 3?
{kind=link}
{kind=link}
Part 4: Transform plasmid from yeast into E. coli
Motivation: Yeast replicate plasmid DNA in low numbers and plasmid DNA is hard to analyze in low concentrations. However, E. coli are engineered to replicate plasmid DNA at high numbers and allow for the easy purification of single scFv plasmids in high concentrations.
Competent cells are fragile and should be handled gently, specifically kept cold and not vortexed.
- Label a 1.5 mL tubes with your team information and chill it in your ice bucket.
- Carefully aliquot 25uL of competent NEB 5-alpha E. coli cells from the front laboratory bench in to your cold tube on ice.
- Add 100ng of the yeast plasmid DNA to the competent bacteria.
- Gently tap the tube with your fingertip 5 times to mix.
- Remember: it is important to keep the competent cells cold. Also, avoid over pipetting and vortexing!
- Incubate your transformation mix on ice for 30 min.
- Carry your ice bucket with your transformation to the heat block at the front laboratory bench.
- Be sure you also take your timer.
- Transfer the tube with your transformation to the heat block set to 42 °C and incubate for exactly 30 sec.
- Remove your transformation from the heat block and immediately put them back in the ice bucket, then incubate for 5 min.
- Pipet 500 uL of pre-warmed SOC media into the transformation.
- Move your transformation to the 37 °C incubator and carefully place them on the nutator.
- Incubate transformation for 1 h.
- Retrieve your transformation from the incubator and alert the teaching faculty that you are ready to plate your samples.
- Plate 100μL of the transformation onto an appropriately labeled LB+Amp agar plate.
- The teaching faculty will demonstrate how you should spread the transformation onto the LB+Amp agar plate by using an ethanol burner to sterilize a glass cell spreader.
- Move your spread plate to the 37 °C incubator where they will incubate overnight.
In your laboratory notebook, complete the following:
- Which gene from pCTcon2 will be most essential for survival of the bacterial that have taken up the scFv plasmid?
- How many plasmids do we estimate will go into each bacterial cell? Explain.
Part 5: Edit homework assignment
Using the pointers from the Comm Lab workshop, edit the figure you prepared for the homework assignment due today. You can submit the updated figure by 10 pm tonight for grading. You will get a grade for submitting the original figure on-time and the updated figure will be graded for content. If you choose not to update your homework, the figure already submitted will be graded for content.
Reagents
- Zymoprep Yeast Plasmid Miniprep II (from Zymo Research)
- Zymolyase
- solution 1
- solution 2
- solution 3
- wash buffer
- SOC medium: 2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, and 20 mM glucose
- LB+Amp plates
- Luria-Bertani (LB) broth: 1% tryptone, 0.5% yeast extract, and 1% NaCl
- Plates prepared by adding 1.5% agar and 100 μg/mL ampicillin (Amp) to LB
Next day: Purify and sequence clone plasmids
Previous day: Enrich candidate clones from library using FACS